
Composición:
Solicitud:
Usado en tratamiento:
Revisión médica por Oliinyk Elizabeth Ivanovna Última actualización de farmacia el 26.06.2023

¡Atención! ¡La información en la página es solo para profesionales médicos! ¡La información se recopila en Fuentes abiertas y puede contener errores significativos! ¡Tenga cuidado y vuelva a verificar toda la información de esta página!
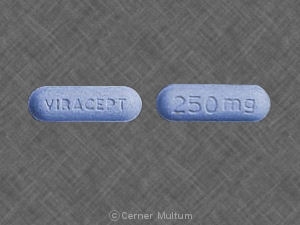
Formas de dosificación y fortalezas
VIRACEPT 250 mg Tableta: Azul claro, en forma de cápsula tabletas con un recubrimiento de película transparente grabado con "VIRACEPT" en un lado y "250" mg "en el otro.
VIRACEPT 625 mg Tableta: tableta ovalada blanca con un transparente recubrimiento de película grabado con "V" en un lado y "625" en el otro.
VIRACEPT Oral Powder: polvo blanquecino que contiene 50 mg (como base libre de nelfinavir) en cada nivel scoopful (1 gramo).
Almacenamiento y manejo
VIRACEPT (mesilato de nelfinavir) 250 mg: Comprimidos de color azul claro en forma de cápsula con un recubrimiento de película transparente grabado con "VIRACEPT" en un lado y "250 mg" en el otro.
Botellas de 300 (250 mg) tabletas - NDC 63010-010-30
VIRACEPT (mesilato de nelfinavir) 625 mg: Comprimido ovalado blanco con una capa de película transparente grabada con "V" por un lado y "625" por el otro.
Botellas de 120 (625 mg) tabletas - NDC 63010-027-70
VIRACEPT (mesilato de nelfinavir) Polvo oral está disponible en polvo blanquecino de 50 mg / g que contiene 50 mg (as base libre de nelfinavir) en cada nivel scoopful (1 gramo).
Botellas de uso múltiple de 144 gramos de polvo con cuchara ...NDC 63010-011-90
VIRACEPT tabletas y orales el polvo debe almacenarse a una temperatura de 15 ° a 30 ° C (59 ° a 86 ° F).
Mantenga el contenedor bien cerrado. Dispense en contenedor original.
Distribuido por: ViiV Healthcare Empresa, Research Triangle Park, NC 27709. Revisado: marzo de 2015
VIRACEPT en combinación con otros agentes antirretrovirales está indicado para el tratamiento de la infección por VIH-1.
Adultos y adolescentes (13 años y mayores)
La dosis recomendada es de 1250 mg (cinco tabletas de 250 mg o dos tabletas de 625 mg) dos veces al día o 750 mg (tres tabletas de 250 mg) tres veces diario. VIRACEPT debe tomarse con una comida. Pacientes incapaces de tragar los 250 o tabletas de 625 mg pueden disolver las tabletas en una pequeña cantidad de agua.
Pacientes pediátricos (de 2 a menos de 13 años)
En niños de 2 años de edad y mayores, lo recomendado la dosis oral de VIRACEPT Oral Powder o tabletas de 250 mg es de 45 a 55 mg / kg dos veces diariamente o de 25 a 35 mg / kg tres veces al día. Todas las dosis deben tomarse con a comida. No se han recibido dosis superiores a la dosis máxima para adultos de 2500 mg por día estudiado en niños.
Para niños que no pueden tragar tabletas, VIRACEPT 250 mg las tabletas pueden disolverse en una pequeña cantidad de agua o, VIRACEPT Oral Powder puede ser administrado.
El proveedor de atención médica debe evaluar lo apropiado formulación y dosificación para cada paciente. Las tablas 1 y 2 proporcionan dosificación Pautas para tabletas y polvo VIRACEPT basados en la edad y el peso corporal.
Tabla 1: Tabla de dosificación para niños de 2 a menos de 13 años
años de edad (tabletas)
| Peso corporal Kg | Dos veces al día (BID) 45 - 55 mg / kg ≥ 2 años | Tres veces al día (TID) 25 - 35 mg / kg ≥ 2 años |
| Número de tabletas (250 mg) | Número de tabletas (250 mg) | |
| 10-12 | 2 | 1 |
| 13-18 | 3 | 2 |
| 19-20 | 4 | 2 |
| ≥ 21 | 4 - 5 * | 3† |
| * Para la dosificación BID, la dosis máxima por día es de 5 tabletas BID † Para la dosificación TID, la dosis máxima por día es de 3 tabletas TID |
Tabla 2: Tabla de dosificación para niños de 2 a menos de 13 años
años de edad (polvo)
| Peso corporal Kg | Dos veces al día (BID) 45 - 55 mg / kg | Tres veces al día (TID) 25 - 35 mg / kg | ||
| cucharadas de polvo (50 mg / 1 g) | Tesoros * de polvo | cucharadas de polvo (50 mg / 1 g) | Tesoros * de polvo | |
| 9.0 a <10.5 | 10 | 2½ | 6 | 1½ |
| 10.5 a <12 | 11 | 2 3/4 | 7 | 1 3/4 |
| 12 a <14 | 13 | 3 1/4 | 8 | 2 |
| 14 a <16 | 15 | 3% | 9 | 2% |
| 16 a <18 | No recomendado † | No recomendado † | 10 | 2½ |
| 18 a <23 | No recomendado † | No recomendado † | 12 | 3 |
| ≥ 23 | No recomendado † | No recomendado † | 15 | 3 3/4 |
| * Si se usa una cucharadita para medir el polvo oral VIRACEPT,
1 cucharadita de nivel contiene 200 mg de VIRACEPT (4 cucharadas de nivel equivalen a 1 nivel
cucharadita) † Use VIRACEPT 250 mg comprimido |
Método de administración
Para pacientes que no pueden tragar tabletas de viracept
- Coloque las tabletas VIRACEPT en una pequeña cantidad de agua.
- Una vez disuelto, mezcle bien el líquido turbio y consuma de inmediato.
- El vidrio debe enjuagarse con agua y enjuagarse tragado para garantizar que se consuma la dosis completa.
Viracept Oral Powder
- Mezcle VIRACEPT Oral Powder con una pequeña cantidad de agua leche, fórmula, fórmula de soya, leche de soya o suplementos dietéticos
- Una vez mezclado, todo el contenido debe consumirse en orden para obtener la dosis completa.
- Si la mezcla no se consume inmediatamente, debe serlo almacenado bajo refrigeración, pero el almacenamiento no debe exceder las 6 horas.
- Alimentos ácidos o jugos (p. Ej., jugo de naranja, jugo de manzana, o salsa de manzana) no se recomiendan para mezclar VIRACEPT Oral Powder porque el La combinación puede dar como resultado un sabor amargo.
- VIRACEPT Oral Powder no debe reconstituirse con agua en su recipiente original.
Insuficiencia hepática
VIRACEPT puede usarse en pacientes con insuficiencia hepática leve deterioro sin ningún ajuste de dosis. VIRACEPT no debe usarse en pacientes con insuficiencia hepática moderada o grave.
La administración conjunta de VIRACEPT está contraindicada con medicamentos que dependen en gran medida del CYP3A para el aclaramiento y para los cuales son elevados Las concentraciones plasmáticas están asociadas con graves y / o potencialmente mortales eventos. Estas drogas y otras drogas contraindicadas (que pueden conducir a una reducción eficacia de nelfinavir) se enumeran en la Tabla 3 [ver también INTERACCIONES DE DROGAS, Tabla 6].
Tabla 3: Drogas contraindicadas con VIRACEPT
| Clase de drogas | Drogas dentro de la clase que están contraindicadas con VIRACEPT | Comentario clínico |
| Antagonista del receptor alfa 1-adrenoreceptor | Alfuzosina | El aumento potencial de las concentraciones de alfuzosina puede provocar hipotensión. |
| Antiarrítmicos | Amiodarona, quinidina | Potencial de arritmia cardíaca grave y / o potencialmente mortal. |
| Agentes antimicobacterianos | Rifampina | Las concentraciones plasmáticas de nelfinavir pueden reducirse mediante el uso concomitante de rifampicina. Esto puede conducir a la pérdida del efecto terapéutico y al posible desarrollo de resistencia a VIRACEPT u otros agentes antirretrovirales administrados conjuntamente. |
| Derivados de ergot | Dihidroergotamina, ergotamina, metilergonovina | Potencial de reacciones graves y / o potencialmente mortales, como la toxicidad del cornezuelo de centeno caracterizada por vasoespasmo periférico e isquemia de las extremidades y otros tejidos. |
| Agente de Motilidad GI | Cisapride | Potencial de reacciones graves y / o potencialmente mortales, como arritmias cardíacas. |
| Productos herbales | S t. Hierba de John (Hypericum perforatum) | Las concentraciones plasmáticas de nelfinavir pueden reducirse mediante el uso concomitante de la preparación a base de hierbas St. La hierba de John. Esto puede conducir a la pérdida del efecto terapéutico y al posible desarrollo de resistencia a VIRACEPT u otros agentes antirretrovirales administrados conjuntamente. |
| Inhibidores de la HMG-CoA Reductasa | Lovastatina, Simvastatina | Potencial de reacciones graves como la miopatía, incluida la rabdomiólisis. |
| Neurolépticos | Pimozida | Potencial de reacciones graves y / o potencialmente mortales, como arritmias cardíacas. |
| Inhibidores de PDE5 | Sildenafil (Revatio®) [para el tratamiento de la hipertensión arterial pulmonar]a | No se ha establecido una dosis segura y efectiva cuando se usa con nelfinavir. Existe un mayor potencial para eventos adversos asociados con el sildenafil (que incluyen trastornos visuales, hipotensión, erección prolongada y síncope). |
| Sedante / Hipnótico s | Triazolam, midazolam oral | Potencial de reacciones graves y / o potencialmente mortales, como sedación prolongada o aumentada o depresión respiratoria. |
| a Ver INTERACCIONES DE DROGAS, Tabla 6 para administración conjunta de sildenafil y tadalafil cuando se dosifica para eréctil disfunción. |
ADVERTENCIAS
Incluido como parte de la PRECAUCIONES sección.
PRECAUCIONES
ALERTA: Conozca los medicamentos que no deberían ser tomado con VIRACEPT . Esta declaración está incluida en la etiqueta de la botella del producto.
Riesgo de reacciones adversas graves debido a interacciones con medicamentos
Iniciación de VIRACEPT, un inhibidor de CYP3A, en pacientes recibir medicamentos metabolizados por CYP3A o inicio de medicamentos metabolizados por CYP3A en pacientes que ya reciben VIRACEPT, puede aumentar el plasma concentraciones de medicamentos metabolizados por CYP3A. Iniciación de medicamentos que inhibe o induce CYP3A puede aumentar o disminuir las concentraciones de VIRACEPT, respectivamente. Estas interacciones pueden conducir a:
- Reacciones adversas clínicamente significativas, potencialmente que conduce a eventos severos, potencialmente mortales o fatales por mayores exposiciones de medicamentos concomitantes.
- Reacciones adversas clínicamente significativas de mayores exposiciones de VIRACEPT .
- Pérdida del efecto terapéutico de VIRACEPT y posible desarrollo de resistencia.
Consulte la Tabla 6 para conocer los pasos para prevenir o administrar estos posibles y interacciones farmacológicas significativas conocidas, incluidas las recomendaciones de dosificación. Considere el potencial de interacciones farmacológicas antes de y durante la terapia VIRACEPT; revisar medicamentos concomitantes durante VIRACEPT terapia; y controlar las reacciones adversas asociadas con el concomitante medicamentos.
Insuficiencia hepática
VIRACEPT no debe usarse en pacientes con ninguno de los dos insuficiencia hepática moderada o grave (Child-Pugh B o C, puntuación mayor que o igual a 7).
Fenilcetonúricos
Viracept Oral Powder contiene fenilalanina, un componente de aspartamo. Cada gramo de polvo VIRACEPT contiene 11,2 mg de fenilalanina. La fenilalanina puede ser perjudicial para los pacientes con fenilcetonuria.
Diabetes mellitus / hiperglucemia
Diabetes mellitus de nueva aparición, exacerbación de preexistente Se han informado diabetes mellitus e hiperglucemia durante la vigilancia posterior a la comercialización en pacientes infectados por el VIH que reciben terapia con inhibidores de la proteasa. Algunos pacientes requirió ajustes de inicio o dosis de insulina o hipoglucemiante oral agentes para el tratamiento de estos eventos. En algunos casos, la cetoacidosis diabética tiene ocurrió. En aquellos pacientes que descontinuaron la terapia con inhibidores de la proteasa, la hiperglucemia persistió en algunos casos. Porque estos eventos han sido reportados voluntariamente durante la práctica clínica, no se pueden hacer estimaciones de frecuencia y una relación causal entre la terapia con inhibidores de la proteasa y estos eventos tiene No se ha establecido.
Hemofilia
Ha habido informes de aumento de sangrado, incluido hematomas cutáneos espontáneos y hemartrosis, en pacientes con hemofilia tipo A y B tratado con inhibidores de la proteasa. En algunos pacientes, factor adicional VIII fue dado. En más de la mitad de los casos reportados, tratamiento con los inhibidores de la proteasa continuaron o se reintrodujeron. Una relación causal tiene No se ha establecido.
Redistribución gorda
Redistribución / acumulación de grasa corporal, incluida la central obesidad, agrandamiento de la grasa dorsocervical ("joroba de búfalo"), desgaste periférico, facial se han observado desperdicios, agrandamiento de los senos y "apariencia cushingoide" pacientes que reciben terapia antirretroviral. El mecanismo y las consecuencias a largo plazo de estos eventos son actualmente desconocidos. Una relación causal no ha sido establecido.
Síndrome de Reconstitución Inmune
Se ha informado el síndrome de reconstitución inmune en pacientes tratados con terapia antirretroviral combinada, incluido VIRACEPT. Durante la fase inicial del tratamiento antirretroviral combinado, pacientes de los cuales El sistema inmunitario responde puede desarrollar una respuesta inflamatoria a indolente o infecciones oportunistas residuales [como la infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jiroveci (PCP) o tuberculosis], que puede requerir una evaluación y tratamiento adicionales.
Trastornos autoinmunes (como la enfermedad de Graves) También se ha informado que la polimiositis y el síndrome de Guillain-Barré) ocurren en el establecimiento de la reconstitución inmune; sin embargo, el tiempo de inicio es mayor variable, y puede ocurrir muchos meses después del inicio del tratamiento.
Información de asesoramiento del paciente
Ver Paciente aprobado por la FDA etiquetado (INFORMACIÓN DE PACIENTES)
Una declaración a los pacientes y los proveedores de atención médica están incluidos en la etiqueta del frasco del producto: ALERTA: Descúbrelo sobre medicamentos que NO deben tomarse con VIRACEPT .
- Instrucción de uso
Para una absorción óptima, Se debe aconsejar a los pacientes que tomen VIRACEPT con alimentos.
Los pacientes deben ser informados que las tabletas VIRACEPT están recubiertas con película y que este recubrimiento de película está destinado a hacerlo hacer que las tabletas sean más fáciles de tragar.
Si es una dosis de VIRACEPT omitido, los pacientes deben tomar la dosis lo antes posible y luego regresar a ella su horario normal. Sin embargo, si se omite una dosis, el paciente no debe duplica la siguiente dosis.
Pacientes adultos o pediátricos no poder tragar las tabletas puede disolver las tabletas en una pequeña cantidad de agua:
- Coloque las tabletas VIRACEPT en pequeña cantidad de agua
- Una vez disuelto, mezcla el turbio bien líquido y consumirlo de inmediato.
- El vidrio debe enjuagarse agua y enjuague tragados para garantizar que se consuma la dosis completa
Pacientes pediátricos incapaces de hacerlo las tabletas de golondrina también pueden usar la formulación en polvo:
- Mezcle VIRACEPT Oral Powder con a pequeña cantidad de agua, leche, fórmula, fórmula de soya, leche de soya o suplementos dietéticos
- Una vez mezclado, todo el contenido debe consumirse para obtener la dosis completa.
- Si la mezcla no se consume inmediatamente, debe almacenarse bajo refrigeración, pero el almacenamiento no debe exceder 6 horas.
- Alimentos ácidos o jugos (p. Ej., jugo de naranja, jugo de manzana o salsa de manzana) no se recomiendan para mezclar VIRACEPT Oral Powder porque la combinación puede provocar un sabor amargo.
- VIRACEPT Oral Powder no debe ser reconstituido con agua en su recipiente original.
Interacciones farmacológicas
VIRACEPT puede interactuar con algunos drogas; por lo tanto, se debe aconsejar a los pacientes que informen a su médico sobre el uso de cualquier otro medicamento recetado, sin receta o productos herbales particularmente St. Hierba de John.
Pacientes que reciben oral los anticonceptivos deben ser instruidos como anticonceptivos alternativos o adicionales Se deben usar medidas durante la terapia con VIRACEPT
Pacientes que reciben sildenafil u otros inhibidores de PDE5, y se debe informar a nelfinavir que pueden estar en un mayor riesgo de eventos adversos asociados con el inhibidor de PDE5, incluidos hipotensión, cambios visuales y erección del pene prolongada, y debe ser puntual informar cualquier síntoma a su médico.
Insuficiencia hepática
Los pacientes deben ser informados que VIRACEPT no debe usarse si hay hepático moderado o grave deterioro.
Fenilcetonuria
Los médicos deben alertar pacientes con fenilcetonuria que VIRACEPT Oral Powder contiene fenilalanina
Redistribución gorda
Los pacientes deben ser informados que la redistribución o acumulación de grasa corporal puede ocurrir en pacientes que reciben terapia antirretroviral, incluida PREZISTA / ritonavir, y que la causa y Los efectos a largo plazo en la salud de estas afecciones no se conocen en este momento
El evento adverso más frecuente asociado con VIRACEPT es la diarrea, que generalmente se puede controlar con ella medicamentos sin receta, como la loperamida, que disminuyen el gastrointestinal motilidad.
Información general
VIRACEPT no es una cura para La infección por VIH-1 y los pacientes pueden continuar experimentando enfermedades asociadas con infección por VIH-1, incluidas infecciones oportunistas. Los pacientes deben permanecer bajo el cuidado de un médico cuando use VIRACEPT. Los pacientes deben estarlo aconsejó evitar hacer cosas que puedan propagar la infección por VIH-1 a otros.
- No compartir agujas o otro equipo de inyección.
- No comparta artículos personales que puede tener sangre o fluidos corporales, como cepillos de dientes y cuchillas de afeitar.
- No tengo ningún tipo de sexo sin protección. Siempre practica con seguridad sexo usando un condón de látex o poliuretano para reducir la posibilidad de contacto sexual con semen, secreciones vaginales o sangre.
- No amamante. No sabemos si VIRACEPT puede pasar a su bebé su leche materna y si podría dañar a su bebé. Además, madres con VIH-1 no debe amamantar porque el VIH-1 puede transmitirse al bebé en el seno leche.
Se debe decir eso a los pacientes Las disminuciones sostenidas en el ARN del VIH en plasma se han asociado con un riesgo reducido de progresión al SIDA y muerte.
Los pacientes deben permanecer debajo el cuidado de un médico mientras usa VIRACEPT. Se debe aconsejar a los pacientes que lo hagan tome VIRACEPT y otra terapia antirretroviral concomitante todos los días como prescrito. Los pacientes no deben alterar la dosis o suspender la terapia sin consultar con su doctor.
Toxicología no clínica
Carcinogénesis, mutagénesis, deterioro de la fertilidad
Se realizaron estudios de carcinogenicidad en ratones y ratas con nelfinavir a dosis orales de hasta 1000 mg / kg / día. No hay evidencia de una tumorigénica se observó efecto en ratones con exposiciones sistémicas (Cmax) hasta 9 veces más medido en humanos a la dosis terapéutica recomendada (750 mg TID o 1250 mg BID). En ratas, se incrementaron los adenomas y carcinomas de células foliculares tiroideas en hombres a 300 mg / kg / día y más y en mujeres a 1000 mg / kg / día. Sistémico las exposiciones (Cmax) a 300 y 1000 mg / kg / día fueron de 1 a 3 veces, respectivamente los medidos en humanos a la dosis terapéutica recomendada. Repetido La administración de nelfinavir a ratas produjo efectos consistentes con el hepático inducción enzimática microsómica y aumento de la deposición de hormona tiroidea; estos los efectos predisponen a las ratas, pero no a los humanos, a las neoplasias de células foliculares tiroideas. El nelfinavir no mostró evidencia de actividad mutagénica o clastogénica en una batería de in vitro y in vivo ensayos de toxicología genética. Estos estudios incluidos ensayos de mutación bacteriana en S. typhimurium y E. coli, un ratón ensayo de linfoma tirosina quinasa, un ensayo de aberración cromosómica en humanos linfocitos, y an in vivo ensayo de micronúcleos de médula ósea de ratón.
El nelfinavir no produjo efectos ni en hombres ni en mujeres apareamiento y fertilidad o supervivencia embrionaria en ratas a exposiciones sistémicas comparables a la exposición terapéutica humana.
Uso en poblaciones específicas
Embarazo
Embarazo Categoría B
VIRACEPT debe usarse durante el embarazo solo si el El beneficio potencial justifica el riesgo potencial para el feto. No hay estudios adecuados y bien controlados en mujeres embarazadas que toman VIRACEPT
No hubo efectos sobre el desarrollo fetal o materno toxicidad cuando se administró nelfinavir a ratas preñadas con exposiciones sistémicas (AUC) comparable a la exposición humana. Administración de nelfinavir a embarazadas los conejos no tuvieron efectos de desarrollo fetal hasta una dosis a la que un poco se observó disminución en el peso corporal materno; sin embargo, incluso en el más alto dosis evaluada, la exposición sistémica en conejos fue significativamente menor que la humana exposición. Estudios adicionales en ratas indicaron que la exposición al nelfinavir en las mujeres desde mediados del embarazo hasta la lactancia no tuvieron ningún efecto sobre la supervivencia crecimiento y desarrollo de la descendencia al destete. Reproductivo posterior El rendimiento de estos descendientes tampoco se vio afectado por la exposición materna a nelfinavir.
Registro de embarazo antirretroviral (APR)
Monitorear los resultados materno-fetales de las mujeres embarazadas expuesto a VIRACEPT y otros agentes antirretrovirales, un antirretroviral Se ha establecido el Registro de embarazo. Se alienta a los médicos a registrarse pacientes llamando al (800) 258-4263.
Madres lactantes
Los Centros para el Control y la Prevención de Enfermedades recomienda que las madres infectadas por el VIH no amamanten a sus bebés para evitar riesgos transmisión postnatal del VIH Se han demostrado estudios en ratas lactantes que nelfinavir se excreta en la leche. Debido a ambos, el potencial para el VIH transmisión y el potencial de reacciones adversas graves en lactantes, se debe indicar a las madres que no amamanten si están recibiendo VIRACEPT
Uso pediátrico
La seguridad, la tolerabilidad, el perfil farmacocinético y La eficacia de VIRACEPT se evaluó en pacientes pediátricos infectados por VIH de 2 a 13 años de edad en ensayos clínicos multicéntricos, Estudio 556 y PACTG 337. En pacientes menores de 2 años edad, VIRACEPT se encontró seguro a las dosis estudiadas, pero de manera confiable no se pudo establecer una dosis efectiva. El perfil farmacocinético seguridad y actividad antiviral de VIRACEPT en pacientes adolescentes de 13 años y más antiguo está respaldado por datos de ensayos clínicos para adultos donde algunos ensayos inscripción permitida de asignaturas de 13 años o más. Por lo tanto, los datos para adolescentes y adultos fueron analizados colectivamente.
Uso geriátrico
Los estudios clínicos de VIRACEPT no incluyeron suficientes cantidad de sujetos de 65 años o más para determinar si responden de manera diferente de sujetos más jóvenes.
Insuficiencia hepática
VIRACEPT no debe usarse en pacientes con ninguno de los dos insuficiencia hepática moderada o grave (Child-Pugh B o C, puntuación mayor que o igual a 7). No es necesario ajustar la dosis de VIRACEPT en pacientes con insuficiencia hepática leve deterioro (Child-Pugh A, puntaje 5-6).
Deterioro renal
La seguridad y eficacia de VIRACEPT no lo han sido establecido en pacientes con insuficiencia renal.
CYP3A y CYP2C19 parecen ser las enzimas predominantes que metabolizan nelfinavir en humanos. La capacidad potencial de nelfinavir para inhibir las principales enzimas humanas del citocromo P450 (CYP3A, CYP2C19, CYP2D6, CYP2C9, CYP1A2 y CYP2E1) ha sido investigado in vitro Solo CYP3A fue. inhibido a concentraciones en el rango terapéutico. Interacción farmacológica específica Se realizaron estudios con nelfinavir y varios medicamentos. La tabla 12 resume los efectos de nelfinavir en el AUC, Cmax y Cmin medios geométricos de drogas administradas conjuntamente. La Tabla 13 muestra los efectos de los medicamentos administrados conjuntamente en el AUC, Cmax y Cmin medias geométricas de nelfinavir.
Tabla 12: Interacciones farmacológicas: cambios en la farmacocinética Parámetros para el medicamento administrado conjuntamente en presencia de VIRACEPT
| Droga coadministrada | Dosis de nelfinavir | N | % Cambio de parámetros farmacocinéticos de medicamentos administrados * (IC 90%) | ||
| AUC | Cmax | Cmin | |||
| Inhibidores de la proteasa del VIH | |||||
| Indinavir 800 mg dosis única | 750 mg q8h x 7 días | 6 | ↑ 51% (↑ 29- ↑ 77%) |
↓ 10% (↓ 28- ↑ 13%) |
NA |
| Ritonavir 500 mg dosis única | 750 mg q8h x 5 dosis | 10 | ↔ | ↔ | NA |
| Saquinavir 1200 mg dosis única † | 750 mg TID x 4 días | 14 | ↑ 392% (↑ 291- ↑ 521%) |
↑ 179% (↑ 117- ↑ 259%) |
NA |
| Amprenavir 800 mg TID x 14 días | 750 mg TID x 14 días | 6 | ↔ | ↓ 14% (↓ 38- ↑ 20%) |
↑ 189% (↑ 52- ↑ 448%) |
| Inhibidores de la transcriptasa inversa de nucleósidos | |||||
| Lamivudina 150 mg dosis única | 750 mg q8h x 7-10 días | 11 | ↑ 10% (↑ 2- ↑ 18%) |
↑ 31% (↑ 9- ↑ 56%) |
NA |
| Zidovudina 200 mg dosis única | 750 mg q8h x 7-10 días | 11 | ↓ 35% (↓ 29- ↓ 40%) |
↓ 31% (↓ 13- ↓ 46%) |
NA |
| Inhibidores de la transcriptasa inversa no nucleósidos | |||||
| Efavirenz 600 mg qd x 7 días | 750 mg q8h x 7 días | 10 | ↓ 12% (↓ 31- ↑ 12%) |
↓ 12% (↓ 29- ↑ 8%) |
↓ 22% (↓ 54- ↑ 32%) |
| Delavirdina 400 mg q8h x 14 días | 750 mg q8h x 7 días | 7 | ↓ 31% (↓ 57- ↑ 10%) |
↓ 27% (↓ 49- ↑ 4%) |
↓ 33% (↓ 70- ↑ 49%) |
| Agentes antiinfecciosos | |||||
| Rifabutina 150 mg qd x 8 días§ | 750 mg q8h x 7-8 días 1 | 12 | ↑ 83% (↑ 72- ↑ 96%) |
↑ 19% (↑ 11- ↑ 28%) |
↑ 177% (↑ 144- ↑ 215%) |
| Rifabutina 300 mg qd x 8 días | 750 mg q8h x 7-8 días | 10 | ↑ 207% (↑ 161- ↑ 263%) |
↑ 146% (↑ 118- ↑ 178%) |
↑ 305% (↑ 245- ↑ 375%) |
| Azitromicina 1200 mg Dosis única | 750 mg TID x 11 días | 12 | ↑ 112% (↑ 80- ↑ 150%) |
↑ 136% (↑ 77- ↑ 215%) |
NA |
| Inhibidores de la HMG-CoA Reductasa | |||||
| Atorvastatina 10 mg qd x 28 días | 1250 mg BID x 14 días | 15 | ↑ 74% (↑ 41- ↑ 116%) |
↑ 122% (↑ 68- ↑ 193%) |
↑ 39% (↓ 21- ↑ 145%) |
| Simvastatina 20 mg qd x 28 días | 1250 mg BID x 14 días | 16 | ↑ 505% (↑ 393- ↑ 643%) |
↑ 517% (↑ 367- ↑ 715%) |
ND |
| Otros agentes | |||||
| Etinilestradiol 35 | ag qd x 15 días | 750 mg q8h x 7 días | 12 | ↓ 47% (↓ 42- ↓ 52%) |
↓ 28% (↓ 16- ↓ 37%) |
↓ 62% (↓ 57- ↓ 67%) |
| Noretindrona 0.4 mg qd x 15 días | 750 mg q8h x 7 días | 12 | ↓ 18% (↓ 13- ↓ 23%) |
↔ | ↓ 46% (↓ 38- ↓ 53%) |
| Metadona 80 mg ± 21 mg qd #> 1 mes | 1250 mg BID x 8 días | 13 | ↓ 47% (↓ 42- ↓ 51%) |
↓ 46% (↓ 42- ↓ 49%) |
↓ 53% (↓ 49- ↓ 57%) |
| Fenitoína 300 mg qd x 14 díasÞ | 1250 mg BID x 7 días | 12 | ↓ 29% (↓ 17- ↓ 39%) |
↓ 21% (↓ 12- ↓ 29%) |
↓ 39% (↓ 27- ↓ 49%) |
| NA: No es relevante para el tratamiento de dosis única; ND: no puede ser
determinado * ↑ Indica aumento; ↓ Indica disminución; ↔ Indica no cambio (la exposición media geométrica aumentó o disminuyó <10%) † Usando la formulación de cápsulas de gelatina blanda de saquinavir 1200 mg § Los cambios de qd de rifabutina 150 mg son relativos a 300 mg de qd x 8 días de rifabutina sin administración conjunta con nelfinavir Se observaron cambios comparables en las concentraciones de rifabutina con VIRACEPT 1250 mg q12h x 7 días # Se informan cambios para la metadona plasmática total; cambios para el individuo El enantiómero R y el enantiómero S fueron similares Se informan medidas de exposición a fenitoína para la exposición total a fenitoína. Los El efecto de nelfinavir sobre la fenitoína no unida fue similar |
|||||
Tabla 13: Interacciones farmacológicas: cambios en
Parámetros farmacocinéticos para nelfinavir en presencia de la administración conjunta
Droga
| Droga coadministrada | Dosis de nelfinavir | N | % Cambio de parámetros farmacocinéticos de nelfinavir * (IC 90%) | ||
| AUC | C max | C min | |||
| Inhibidores de la proteasa del VIH | |||||
| Indinavir 800 mg q8h x 7 días | 750 mg dosis única | 6 | ↑ 83% (↑ 42- ↑ 137%) |
↑ 31% (↑ 16- ↑ 48%) |
NA |
| Ritonavir 500 mg q12h x 3 dosis | 750 mg dosis única | 10 | ↑ 152% (↑ 96- ↑ 224%) |
↑ 44% (↑ 28- ↑ 63%) |
NA |
| Saquinavir 1200 mg TID x 4 días | 750 mg dosis única | 14 | ↑ 18% (↑ 7- ↑ 30%) |
↔ | NA |
| Inhibidores de la transcriptasa inversa de nucleósidos | |||||
| Didanosina 200 mg dosis única | 750 mg dosis única | 9 | ↔ | ↔ | NA |
| Zidovudina 200 mg + Lamivudina 150 mg Dosis única | 750 mg q8h x 7-10 días | 11 | ↔ | ↔ | ↔ |
| Inhibidores de la transcriptasa inversa no nucleósidos | |||||
| Efavirenz 600 mg qd x 7 días | 750 mg q8h x 7 días | 7 | ↑ 20% (↑ 8- ↑ 34%) |
↑ 21% (↑ 10- ↑ 33%) |
↔ |
| Nevirapina 200 mg qd x 14 días seguido de 200 mg BID x 14 días | 750 mg TID x 36 días | 23 | ↔ | ↔ | ↓ 32% (↓ 50- ↑ 5%) |
| Delavirdina 400 mg q8h x 7 días | 750 mg q8h x 14 días | 12 | ↑ 107% (↑ 83- ↑ 135%) |
↑ 88% (↑ 66- ↑ 113%) |
↑ 136% (↑ 103- ↑ 175%) |
| Agentes antiinfecciosos | |||||
| Ketoconazol 400 mg qd x 7 días | 500 mg q8h x 5-6 días | 12 | ↑ 35% (↑ 24- ↑ 46%) |
↑ 25% (↑ 11- ↑ 40%) |
↑ 14% (↓ 23- ↑ 69%) |
| Rifabutina 150 mg qd x 8 días | 750 mg q8h x 7-8 días | 11 | ↓ 23% (↓ 14- ↓ 31%) |
↓ 18% (↓ 8-↓27%) |
↓ 25% (↓ 8-↓ 39%) |
| 1250 mg q12h x 7-8 días | 11 | ↔ | ↔ | ↓ 15% (↓ 43- ↑ 27%) |
|
| Rifabutina 300 mg qd x 8 días | 750 mg q8h x 7-8 días | 10 | ↓ 32% (↓ 15- ↓ 46%) |
↓ 24% (↓ 10- ↓ 36%) |
↓ 53% (↓ 15- ↓ 73%) |
| Rifampina 600 mg qd x 7 días | 750 mg q8h x 5-6 días | 12 | ↓ 83% (↓ 79- ↓ 86%) |
↓ 76% (↓ 69- ↓ 82%) |
↓ 92% (↓ 86- ↓ 95%) |
| Azitromicina 1200 mg Dosis única | 750 mg tid x 9 días | 12 | ↓ 15% (↓ 7- ↓ 22%) |
↓ 10% (↓ 19- ↑ 1%) |
↓ 29% (↓ 19- ↓ 38%) |
| Otros agentes | |||||
| Fenitoína 300 mg qd x 7 días | 1250 mg BID x 14 días | 15 | ↔ | ↔ | ↓ 18% (↓ 45- ↑ 23%) |
| Omeprazol 40 mg qd x 4 días administrado 30 minutos antes de nelfinavir | 1250 mg BID x 4 días | 19 | ↓ 36% (↓ 20- ↓ 49%) |
↓ 37% (↓ 23- ↓ 49%) |
↓ 39% (↓ 15- ↓ 57%) |
| NA: No es relevante para el tratamiento de dosis única * ↑ Indica aumento; ↓ Indica disminución; ↔ Indica no cambio (la exposición media geométrica aumentó o disminuyó <10%) † Usando la formulación de cápsulas de gelatina blanda de saquinavir 1200 mg |
|||||
Embarazo Categoría B
VIRACEPT debe usarse durante el embarazo solo si el El beneficio potencial justifica el riesgo potencial para el feto. No hay estudios adecuados y bien controlados en mujeres embarazadas que toman VIRACEPT
No hubo efectos sobre el desarrollo fetal o materno toxicidad cuando se administró nelfinavir a ratas preñadas con exposiciones sistémicas (AUC) comparable a la exposición humana. Administración de nelfinavir a embarazadas los conejos no tuvieron efectos de desarrollo fetal hasta una dosis a la que un poco se observó disminución en el peso corporal materno; sin embargo, incluso en el más alto dosis evaluada, la exposición sistémica en conejos fue significativamente menor que la humana exposición. Estudios adicionales en ratas indicaron que la exposición al nelfinavir en las mujeres desde mediados del embarazo hasta la lactancia no tuvieron ningún efecto sobre la supervivencia crecimiento y desarrollo de la descendencia al destete. Reproductivo posterior El rendimiento de estos descendientes tampoco se vio afectado por la exposición materna a nelfinavir.
Registro de embarazo antirretroviral (APR)
Monitorear los resultados materno-fetales de las mujeres embarazadas expuesto a VIRACEPT y otros agentes antirretrovirales, un antirretroviral Se ha establecido el Registro de embarazo. Se alienta a los médicos a registrarse pacientes llamando al (800) 258-4263.
Porque los ensayos clínicos se realizan ampliamente condiciones variables, tasas de reacción adversas observadas en los ensayos clínicos de a el fármaco no se puede comparar directamente con las tasas en los ensayos clínicos de otro droga y puede no reflejar las tasas observadas en la práctica.
Experiencia en ensayos clínicos: adultos y adolescentes (13 Años y mayores)
La seguridad de VIRACEPT se estudió en más de 5000 pacientes que recibieron drogas solo o en combinación con análogos de nucleósidos. Los La mayoría de los eventos adversos fueron de intensidad leve. El más frecuentemente reportado El evento adverso entre los pacientes que recibieron VIRACEPT fue la diarrea, que fue generalmente de intensidad leve a moderada.
Experiencias adversas clínicas relacionadas con medicamentos de moderado o intensidad severa en ≥ 2% de los pacientes tratados con VIRACEPT coadministrado con d4T y 3TC (Estudio 542) por hasta 48 semanas, o con ZDV más 3TC (Estudio 511) hasta 24 semanas se presentan en la Tabla 4.
Tabla 4: Porcentaje de pacientes con
Tratamiento-emergente * Eventos adversos de intensidad moderada o severa informados en ≥
2% de pacientes adultos y adolescentes
| Eventos adversos | Estudio 511 24 semanas | Estudio 542 48 semanas | |||
| Placebo + ZDV / 3TC (n = 101) |
500 mg de VIRACEPT TID + ZDV / 3TC (n = 97) |
750 mg de VIRACEPT TID + ZDV / 3TC (n = 100) |
1250 mg BID VIRACEPT + d4T / 3TC (n = 344) |
750 mg de VIRACEPT TID + d4T / 3TC (n = 210) |
|
| Sistema digestivo | |||||
| Diarrea | 3% | 14% | 20% | 20% | 15% |
| Náuseas | 4% | 3% | 7% | 3% | 3% |
| Flatulencia | 0 | 5% | 2% | 1% | 1% |
| Piel / Apéndices | |||||
| Sarpullido | 1% | 1% | 3% | 2% | 1% |
| * Incluye esos eventos adversos al menos posiblemente probablemente o definitivamente relacionado con el fármaco del estudio o de una relación desconocida y excluye las condiciones concurrentes del VIH | |||||
Eventos adversos que ocurren en menos del 2% de los pacientes recibir VIRACEPT en todos los ensayos clínicos de fase 2 y 3 y considerado al menos posiblemente relacionado o de relación desconocida con el tratamiento y de al menos la gravedad moderada se enumeran a continuación.
Cuerpo en su conjunto: dolor abdominal, accidental lesión, reacción alérgica, astenia, dolor de espalda, fiebre, dolor de cabeza, malestar general, dolor, etc y redistribución / acumulación de grasa corporal.
Sistema digestivo: anorexia, dispepsia, epigástrica dolor, sangrado gastrointestinal, hepatitis, ulceración bucal, pancreatitis y vómitos.
Sistema Hemico / Linfático : anemia, leucopenia y trombocitopenia.
Sistema metabólico / nutricional: aumenta en fosfatasa alcalina, amilasa, creatina fosfoquinasa, deshidrogenasa láctica SGOT, SGPT y gamma-glutamil transpeptidasa; hiperlipemia, hiperuricemia, hiperglucemia, hipoglucemia, deshidratación y pruebas de función hepática anormales.
Sistema musculoesquelético : artralgia, artritis calambres, mialgia, miastenia y miopatía.
Sistema nervioso: ansiedad, depresión, mareos labilidad emocional, hipercinesia, insomnio, migraña, parestesia, convulsiones trastorno del sueño, somnolencia e ideación suicida.
Sistema respiratorio: disnea, faringitis rinitis y sinusitis.
Piel / Apéndices : dermatitis, foliculitis, hongos dermatitis, erupción maculopapular, prurito, sudoración y urticaria.
Sentidos especiales: iritis aguda y trastorno ocular.
Sistema urogenital : cálculo renal, sexual disfunción y anormalidad urinaria.
Anomalías de laboratorio
El porcentaje de pacientes con laboratorio marcado Las anormalidades en los Estudios 542 y 511 se presentan en la Tabla 5. Marcado Las anormalidades de laboratorio se definen como una anormalidad de Grado 3 o 4 en un paciente con un valor basal normal o una anormalidad de grado 4 en un paciente con a Anomalía de grado 1 al inicio del estudio.
Tabla 5: Porcentaje de pacientes por grupo de tratamiento
con anormalidades de laboratorio marcadas * en> 2% de pacientes
| Estudio 511 | Estudio 542 | ||||
| Placebo + ZDV / 3TC (n = 101) |
500 mg TID VIRACEPT + ZDV / 3TC (n = 97) |
750 mg TID VIRACEPT + ZDV / 3TC (n = 100) |
1250 mg BID VIRACEPT + d4T / 3TC (n = 344) |
750 mg TID VIRACEPT + d4T / 3TC (n = 210) |
|
| Hematología | |||||
| Hemoglobina | 6% | 3% | 2% | 0 | 0 |
| Neutrófilos | 4% | 3% | 5% | 2% | 1% |
| Linfocitos | 1% | 6% | 1% | 1% | 0 |
| Química | |||||
| ALT (SGPT) | 6% | 1% | 1% | 2% | 1% |
| AST (SGOT) | 4% | 1% | 0 | 2% | 1% |
| Creatina Kinase | 7% | 2% | 2% | NA | NA |
| * Las anomalías marcadas en el laboratorio se definen como un cambio desde el grado 0 al inicio hasta al menos el grado 3 o desde el grado 1 hasta el grado 4 | |||||
Experiencia en ensayos clínicos: pediatría (2 a menos de 13 Años de edad)
VIRACEPT se ha estudiado en aproximadamente 400 pediátricos pacientes en ensayos clínicos desde el nacimiento hasta los 13 años de edad. El perfil de eventos adversos visto durante ensayos clínicos pediátricos fue similar al de los adultos.
El más comúnmente reportado relacionado con las drogas Los eventos adversos emergentes del tratamiento informados en los estudios pediátricos incluyeron: diarrea, leucopenia / neutropenia, erupción cutánea, anorexia y dolor abdominal. Diarrea, independientemente de la relación asignada al fármaco del estudio, se informó en 39% a 47% de pacientes pediátricos que reciben VIRACEPT en 2 de los ensayos de tratamiento más grandes. La leucopenia / neutropenia fue la anormalidad de laboratorio más comúnmente reportada como Un evento significativo en los estudios pediátricos.
Experiencia de postmarketing
Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de VIRACEPT. Porque estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible de manera confiable estimar su frecuencia o establecer una relación causal con la exposición a drogas.
Cuerpo en su conjunto: reacciones de hipersensibilidad (incluyendo broncoespasmo, erupción cutánea moderada a grave, fiebre y edema).
Sistema cardiovascular: QTc prolongación, torsades de pointes.
Sistema digestivo: ictericia.
Sistema metabólico / nutricional: bilirrubinemia acidosis metabólica.
La experiencia humana de sobredosis aguda con VIRACEPT es limitado. No existe un antídoto específico para la sobredosis con VIRACEPT. Si está indicado, La eliminación del fármaco no absorbido debe lograrse mediante emesis o lavado gástrico. La administración de carbón activado también se puede utilizar para ayudar a eliminar droga no absorbida. Como el nelfinavir está altamente unido a proteínas, la diálisis es poco probable para eliminar significativamente la droga de la sangre.
Efectos sobre el electrocardiograma
El efecto de Viracept a la dosis recomendada de 1250 mg dos veces al día en el intervalo QTcF administrado con una comida baja en grasa (20% de grasa) fue evaluado en aleatorizado, placebo y activo (moxifloxacina 400 mg una vez al día) estudio cruzado controlado en 66 sujetos sanos. La media máxima diferencias de tiempo coincidente (95% límite de confianza superior) en el intervalo QTcF desde placebo después de la corrección basal fue inferior a 10 milisegundos, el umbral de preocupación clínica. Este hallazgo no cambió cuando una dosis supraterapéutica única de Viracept 3125 mg se administró después de una administración de 3 días de Viracept 1250 mg dos veces al día. La exposición a 3125 mg fue 1,4 veces mayor que a 1250 mg. La dosis de 3125 mg en este estudio no logró las exposiciones anticipadas en pacientes que toman una comida rica en grasas (50% de grasa) o con administración concomitante de medicamentos que podrían aumentar la exposición a nelfinavir.
Ningún sujeto en ningún grupo tuvo un aumento en QTcF de ≥ 60 milisegundos línea de base. Ningún sujeto experimentó un intervalo que exceda potencialmente el potencial umbral clínicamente relevante de 500 milisegundos.
Las propiedades farmacocinéticas de nelfinavir fueron evaluado en voluntarios sanos y pacientes infectados por el VIH; No hay diferencias sustanciales fueron observados entre los dos grupos.
Absorción
Parámetros farmacocinéticos de nelfinavir (área bajo el curva de concentración plasmática-tiempo durante un período de 24 horas en estado estacionario [AUC24], concentraciones plasmáticas máximas [Cmáx], concentraciones mínimas de mañana y tarde [Ctrough]) de un estudio farmacocinético en pacientes VIH positivos después de múltiples dosificación con 1250 mg (cinco tabletas de 250 mg) dos veces al día (BID) durante 28 días (10 pacientes) y 750 mg (tres tabletas de 250 mg) tres veces al día (TID) durante 28 días (11 pacientes) se resumen en la Tabla 7.
Tabla 7: Resumen de un estudio farmacocinético en
Pacientes VIH positivos con dosis múltiples de 1250 mg (cinco tabletas de 250 mg) BID
para 28 días y 750 mg (tres tabletas de 250 mg) TID durante 28 días
| Régimen | AUC24 mg • h / L |
Cmax mg / L |
Ctrough Mañana mg / L |
Ctrough Tarde o tarde mg / L |
| 1250 mg BID | 52,8 ± 15,7 | 4.0 ± 0.8 | 2.2 ± 1.3 | 0.7 ± 0.4 |
| 750 mg TID | 43,6 ± 17,8 | 3.0 ± 1.6 | 1.4 ± 0.6 | 1.0 ± 0.5 |
| Los datos son medios ± DE |
La diferencia entre mañana y tarde o tarde También se observaron concentraciones mínimas para los regímenes TID y BID en salud voluntarios que fueron dosificados a intervalos de 8 o 12 horas.
En voluntarios sanos que reciben una dosis única de 1250 mg, la tableta de 625 mg no fue bioequivalente a la formulación de tableta de 250 mg. Debajo condiciones de ayuno (n = 27), el AUC y la Cmáx fueron 34% y 24% más altos respectivamente, para las tabletas de 625 mg. En una evaluación relativa de biodisponibilidad en condiciones de alimentación (n = 28), el AUC fue un 24% mayor para la tableta de 625 mg; la Cmax fue comparable para ambas formulaciones. En sujetos infectados con VIH-1 (N = 21) recibiendo múltiples dosis de 1250 mg BID en condiciones de alimentación, la formulación de 625 mg fue bioequivalente a la formulación de 250 mg basada en la similitud en estado estacionario exposición (Cmax y AUC).
La Tabla 8 muestra el resumen del estado estacionario parámetros farmacocinéticos (media ± DE) de nelfinavir después de la administración de dosis múltiples de 1250 mg BID (2 x 625 mg comprimidos) a pacientes infectados por el VIH (N = 21) durante 14 dias.
Tabla 8: Resumen de la farmacocinética de estado estacionario
Parámetros (media ± DE) de Nelfinavir después de la administración de dosis múltiples de 1250
mg BID (2 x 625 mg comprimidos) a pacientes infectados por VIH (N = 21) durante 14 días.
| Régimen | AUC12 mg • h / L |
Cmax mg / L |
Cmin mg / L |
| 1250 mg BID | 35,3 (16,4) | 4.7 (1.9) | 1.5 (1.0) |
| AUC12: estado estacionario AUC Cmax: concentración plasmática máxima en estado estacionario Cmin: concentración plasmática mínima en estado estacionario |
En voluntarios sanos que reciben una dosis única de 750 mg En condiciones de alimentación, las concentraciones de nelfinavir fueron similares después de la administración de la tableta de 250 mg y polvo oral.
Efecto de los alimentos sobre la absorción oral
Los alimentos aumentan la exposición y disminuyen al nelfinavir variabilidad farmacocinética de nelfinavir en relación con el estado en ayunas. En uno En el estudio, voluntarios sanos recibieron una dosis única de 1250 mg de VIRACEPT 250 mg tabletas (5 tabletas) en ayunas o en condiciones de alimentación (tres comidas diferentes). En En un segundo estudio, voluntarios sanos recibieron dosis únicas de 1250 mg de VIRACEPT (5 x tabletas de 250 mg) en condiciones de ayuno o alimentación (dos contenidos de grasa diferentes comidas). Los resultados de los dos estudios se resumen en la Tabla 9 y la Tabla 10, respectivamente.
Tabla 9: Aumento de AUC, Cmax y Tmax para Nelfinavir
en estado de la Fed relativo al estado de ayuno después de 1250 mg de VIRACEPT (5 x 250 mg
Tabletas)
| Número de Kcal | % Gordo | Número de sujetos | AUC pliegue aumento | Cmax pliegue aumento | Aumento de Tmax (hr) |
| 125) | 20 | n = 21 | 2.2 | 2.0 | 1.00 |
| 500 | 20 | n = 22 | 3.1 | 2.3 | 2.00 |
| 1000 | 50 | n = 23 | 5.2 | 3.3 | 2.00 |
Tabla 10: Aumento de Nelfinavir AUC, Cmax y Tmax en
Fed Low Fat (20%) versus High Fat (50%) State Relativo al Estado Fasted
Después de 1250 mg de VIRACEPT (5 x 250 mg comprimidos)
| Número de Kcal | % Gordo | Número de sujetos | AUC pliegue aumento | Cmax pliegue aumento | Aumento de Tmax (hr) |
| 500 | 20 | n = 22 | 3.1 | 2.5 | 1.8 |
| 500 | 50 | n = 22 | 5.1 | 3.8 | 2.1 |
La exposición al nelfinavir puede aumentarse aumentando la contenido calórico o graso en comidas tomadas con VIRACEPT
No se ha realizado un estudio de efectos alimentarios con el 625 mg comprimido. Sin embargo, basado en una comparación entre estudios (n = 26 alimentados vs. n = 26 en ayunas) después de la administración de una dosis única de nelfinavir 1250 mg, la magnitud de El efecto alimentario para la tableta de 625 mg de nelfinavir parece comparable al de Las tabletas de 250 mg. VIRACEPT debe tomarse con una comida.
Distribución
El volumen aparente de distribución después de oral la administración de nelfinavir fue de 2-7 L / kg. El nelfinavir en suero está ampliamente unido a proteínas (> 98%).
Metabolismo
El nelfinavir sin cambios comprendió el 82-86% del plasma total radiactividad después de una dosis oral única de 750 mg de 14C-nelfinavir. In vitro, múltiple son responsables las enzimas del citocromo P-450, incluidos CYP3A y CYP2C19 metabolismo de nelfinavir. Uno de los principales y varios metabolitos oxidativos menores fueron encontrados en plasma. El principal metabolito oxidativo tiene in vitro antiviral actividad comparable a la droga original.
Eliminación
La vida media terminal en plasma fue típicamente de 3.5 a 5 horas. Se recuperó la mayoría (87%) de una dosis oral de 750 mg que contenía 14C-nelfinavir en las heces; La radiactividad fecal consistió en numerosos metabolitos oxidativos (78%) y nelfinavir inalterado (22%). Solo se recuperó el 1-2% de la dosis orina, de las cuales nelfinavir sin cambios fue el componente principal.

































