
コンポーネント:
治療オプション:
Oliinyk Elizabeth Ivanovna 、薬局による医学的評価、 最終更新日:26.06.2023

アテンション! そのこのページの情報は医療専門家のみを対象としています! その情報が収集したオープン源を含めることが可能である重大な誤差! 注意して、このページ上のすべての情報を再確認してください!
投薬形態と強さ。
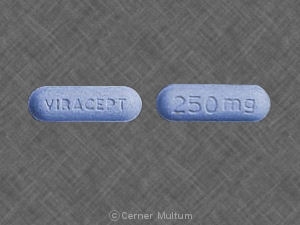
VIRACEPT 250 mg錠剤:水色、カプセル型。 片面に「VIRACEPT」、250が刻印された透明なフィルムコーティングのタブレット。 もう一方のmg」。.
VIRACEPT 625 mg錠剤:透明の白い ⁇ 円形の錠剤。 片面に「V」、面に「625」が刻印されたフィルムコーティング。 その他。.
VIRACEPT経口粉末:50 mgを含むオフホワイト粉末。 (ネルフィナビルフリーベースとして)各レベルのスクープフル(1グラム)。.
保管と取り扱い。
VIRACEPT(メシル酸ネルフィナビル)。 250 mg。:透明なフィルムコーティングが刻印された水色のカプセル型タブレット。 一方に「VIRACEPT」、もう一方に「250 mg」が付いています。.
300(250 mg)錠剤のボトル。 -。 NDC。 63010-010-30。
VIRACEPT(メシル酸ネルフィナビル)。 625 mg。:「V」が刻印された透明なフィルムコーティングが施された白い ⁇ 円形のタブレット。 一方は「625」、もう一方は「625」。.
120(625 mg)錠剤のボトル。 -。 NDC。 63010-027-70。
VIRACEPT(メシル酸ネルフィナビル)。 経口粉末。 50 mgを含む50 mg / gオフホワイトパウダーとして入手できます( ネルフィナビル遊離塩基)各レベルのスクープフル(1グラム)。.
144の複数使用ボトル。 スクープ付きパウダーのグラム……。.NDC。 63010-011-90。
VIRACEPTタブレットと経口。 粉末は15°〜30°C(59°〜86°F)で保管する必要があります。.
容器をしっかりと閉めておいてください。. 元の容器に分注します。.
配布者:ViiV Healthcare。 会社、リサーチトライアングルパーク、ノースカロライナ27709。. 改訂:2015年3月。
他の抗レトロウイルス薬と組み合わせたVIRACEPT。 HIV-1感染症の治療に適応されます。.
大人と青年(13歳以上)。
推奨用量は1250 mg(5つの250 mg錠剤または 2つの625 mg錠剤)を1日2回または750 mg(3つの250 mg錠剤)を3回。 毎日。. VIRACEPTは食事と一緒に服用する必要があります。. 250を飲み込むことができない患者。 または625 mgの錠剤が少量の水に錠剤を溶解する場合があります。.
小児患者(2〜13歳未満)。
2歳以上の子供にお勧めです。 VIRACEPT経口粉末または250 mg錠剤の経口投与量は、45〜55 mg / kgを2回です。 毎日または25〜35 mg / kgを1日3回。. すべての用量はaと一緒に服用する必要があります。 食事。. 成人の最大用量である1日あたり2500 mgを超える用量は、これまで行われていません。 子供たちに勉強した。.
錠剤を飲み込むことができない子供のために、VIRACEPT 250 mg。 錠剤は少量の水またはVIRACEPT経口粉末に溶解できます。 投与することができます。.
医療提供者は適切に評価する必要があります。 各患者の処方と投与量。. 表1および2は投与量を示しています。 年齢と体重に基づくVIRACEPTタブレットとパウダーのガイドライン。.
表1:2〜13歳未満の子供用の投与表。
年齢(タブレット)。
| 体重Kg。 | 1日2回(BID)45-55 mg / kg≥2年。 | 1日3回(TID)25〜35 mg / kg≥2年。 |
| 錠剤の数(250 mg)。 | 錠剤の数(250 mg)。 | |
| 10-12。 | 2 | 1 |
| 13-18。 | 3 | 2 |
| 19-20。 | 4 | 2 |
| ≥21。 | 4-5 *。 | 3† |
| * BID投与の場合、1日あたりの最大用量は5錠BIDです。 ⁇ TID投与の場合、1日あたりの最大用量は3錠TIDです。 |
表2:2〜13歳未満の子供用の投与表。
年齢(粉末)。
| 体重Kg。 | 1日2回(BID)45-55 mg / kg。 | 1日3回(TID)25〜35 mg / kg。 | ||
| 粉末のスクープ(50 mg / 1 g)。 | 粉末の小さじ*。 | 粉末のスクープ(50 mg / 1 g)。 | 粉末の小さじ*。 | |
| 9.0から<10.5。 | 10 | 2½ | 6 | 1½ |
| 10.5から<12。 | 11 | 2 3/4。 | 7 | 1 3/4。 |
| 12から<14。 | 13 | 3 1/4。 | 8 | 2 |
| 14から<16。 | 15 | 3% | 9 | 2% |
| 16から<18。 | 推奨されません ⁇ 。 | 推奨されません ⁇ 。 | 10 | 2½ |
| 18から<23。 | 推奨されません ⁇ 。 | 推奨されません ⁇ 。 | 12 | 3 |
| ≥23。 | 推奨されません ⁇ 。 | 推奨されません ⁇ 。 | 15 | 3 3/4。 |
| *小さじ1杯を使用してVIRACEPT経口粉末を測定する場合。
小さじ1レベルには200 mgのVIRACEPTが含まれています(4レベルのスクープは1レベルに相当します。
小さじ)。 ⁇ VIRACEPT 250 mgタブレットを使用します。 |
投与方法。
ウイルスセプト錠剤を飲み込むことができない患者のため。
- VIRACEPTタブレットを少量の水に入れます。.
- 溶解したら、 ⁇ った液体をよく混ぜて消費します。 すぐに。.
- ガラスは水とすすぎで洗い流してください。 飲み込んで、全量が確実に消費されるようにします。.
ビラセプト経口粉末。
- VIRACEPTオーラルパウダーを少量の水と混ぜます。 牛乳、調合乳、豆乳、豆乳、または栄養補助食品。
- 混合したら、内容物全体を順番に消費する必要があります。 全用量を得るため。.
- 混合物がすぐに消費されない場合は、消費する必要があります。 冷蔵保管されますが、保管は6時間を超えてはなりません。.
- 酸性食品またはジュース(例:.、オレンジジュース、リンゴジュース、または。 アップルソース)は、VIRACEPTオーラルパウダーの混合にはお勧めしません。 組み合わせは苦い味をもたらすかもしれません。.
- VIRACEPTオーラルパウダーは再構成しないでください。 元の容器の水。.
肝障害。
VIRACEPTは、軽度の肝疾患のある患者に使用できます。 用量調整なしの障害。. VIRACEPTは患者に使用しないでください。 中等度または重度の肝機能障害がある。.
VIRACEPTの同時投与は禁 ⁇ です。 クリアランスをCYP3Aに大きく依存し、上昇した薬物。 血漿濃度は、深刻なおよび/または生命を脅かすことに関連しています。 イベント。. これらの薬物およびその他の禁 ⁇ の薬物(これは減少につながる可能性があります。 ネルフィナビルの有効性)を表3に示します[参照]。 薬物相互作用。、。 表6]。.
表3:VIRACEPTで禁 ⁇ の薬物。
| ドラッグクラス。 | VIRACEPTで禁 ⁇ のクラス内の薬物。 | 臨床コメント。 |
| アルファ1-アドレナリン受容体 ⁇ 抗薬。 | アルフゾシン。 | アルフゾシン濃度が上昇する可能性があると、低血圧を引き起こす可能性があります。. |
| 不整脈。 | アミオダロン、キニジン。 | 深刻なおよび/または生命を脅かす心不整脈の可能性。. |
| 抗マイコバクテリア剤。 | リファンピン。 | ネルフィナビルの血漿濃度は、リファンピンを併用することで低減できます。. これは、治療効果の喪失と、VIRACEPTまたは他の同時投与された抗レトロウイルス薬に対する耐性の発生の可能性につながる可能性があります。. |
| エルゴ派。 | ジヒドロエルゴタミン、エルゴタミン、メチルエルゴノビン。 | 末 ⁇ 血管 ⁇ や四肢や他の組織の虚血を特徴とする麦角毒性などの深刻なおよび/または生命を脅かす反応の可能性。. |
| GI Motility Agent。 | シサプリド。 | 心不整脈などの深刻な、および/または生命を脅かす反応の可能性。. |
| ハーブ製品。 | 聖. ジョンの麦 ⁇ (。Hypericum perforatum。) | ネルフィナビルの血漿濃度は、ハーブ製剤St.を併用することで減らすことができます。. ジョンの麦 ⁇ 。. これは、治療効果の喪失と、VIRACEPTまたは他の同時投与された抗レトロウイルス薬に対する耐性の発生の可能性につながる可能性があります。. |
| HMG-CoAレダクターゼ阻害剤。 | ロバスタチン、シンバスタチン。 | 横紋筋融解症を含むミオパシーなどの深刻な反応の可能性。. |
| 神経遮断薬。 | ピモジド。 | 心不整脈などの深刻な、および/または生命を脅かす反応の可能性。. |
| PDE5阻害剤。 | シルデナフィル(Revatio®)[肺動脈高血圧症の治療用]。a | ネルフィナビルと併用した場合、安全で効果的な用量は確立されていません。. シルデナフィルに関連する有害事象(視覚障害、低血圧、勃起の延長、失神など)の可能性が高まっています。. |
| 鎮静/催眠s。 | トリアゾラム、経口ミダゾラム。 | 鎮静または呼吸抑制の長期化または増加などの深刻なおよび/または生命を脅かす反応の可能性。. |
| a 見る。 薬物相互作用。、表6。 勃起のために投与された場合のシルデナフィルとタダラフィルの同時投与。 機能不全。. |
警告。
の一部として含まれています。 注意。 セクション。.
注意。
アラート:すべきではない薬について調べてください。 VIRACEPTで撮影 このステートメントは、製品のボトルラベルに含まれています。.
薬物相互作用による深刻な有害反応のリスク。
CYP3A阻害剤であるVIRACEPTの患者への開始。 CYP3Aによって代謝された薬物の受け取り、または代謝された薬物の開始。 すでにVIRACEPTを受けている患者のCYP3Aにより、血漿が増加する可能性があります。 CYP3Aによって代謝される薬物の濃度。薬の開始。 CYP3Aを阻害または誘導すると、濃度が増加または減少する可能性があります。 それぞれVIRACEPT。. これらの相互作用は以下につながる可能性があります。
- 臨床的に重要な副作用、潜在的に。 より大きな曝露による深刻な、生命を脅かす、または致命的なイベントにつながります。 併用薬。.
- 臨床的に重要な副作用。 VIRACEPTの露出.
- VIRACEPTの治療効果の喪失と可能性。 抵抗の発達。.
これらを防止または管理するための手順については、表6を参照してください。 投与の推奨事項を含む、既知の重要な薬物相互作用。. 前に薬物相互作用の可能性を検討してください。 VIRACEPT療法中; VIRACEPT中の併用薬を確認します。 治療;併用に関連する副作用を監視します。 薬。.
肝障害。
VIRACEPTは、どちらかの患者には使用しないでください。 中等度または重度の肝障害(Child-Pugh BまたはC、スコアがまたはより大きい。 7に等しい)。.
フェニルケトンル。
ビラセプト経口粉末には、成分であるフェニルアラニンが含まれています。 アスパルテームの。. VIRACEPT粉末の各グラムには、11.2 mgのフェニルアラニンが含まれています。. フェニルアラニンはフェニルケトン尿症の患者に害を及ぼす可能性があります。.
糖尿病/高血糖。
新しい発症糖尿病、既存の悪化。 糖尿病と高血糖は市販後調査中に報告されています。 プロテアーゼ阻害剤療法を受けているHIV感染患者。. 一部の患者。 インスリンまたは経口血糖の開始または用量調整のいずれかが必要です。 これらのイベントの治療のための薬剤。. 場合によっては、糖尿病性ケトアシドーシスが持っています。 発生した。. プロテアーゼ阻害剤療法を中止した患者では、 高血糖症が持続する場合もあります。. これらのイベントが報告されているためです。 臨床診療中に自発的に、頻度の推定を行うことはできません。 プロテアーゼ阻害剤療法とこれらのイベントとの因果関係があります。 確立されていません。.
血友病。
出血の増加などの報告があります。 血友病A型患者の自然発生的な皮膚血腫および関節症。 Bプロテアーゼ阻害剤で治療。. 一部の患者では、追加の要因。 VIIIが与えられました。. 報告された症例の半分以上で、治療。 プロテアーゼ阻害剤は継続または再導入されました。. 因果関係があります。 確立されていません。.
脂肪の再分配。
中央を含む体脂肪の再分布/蓄積。 肥満、背 ⁇ 部の脂肪拡大(「バッファローこぶ」)、末 ⁇ 消耗、顔射。 消耗、乳房肥大、「クッシングイドの外観」が観察されています。 抗レトロウイルス療法を受けている患者。. メカニズムと長期的な影響。 これらのイベントの現在不明です。. 因果関係はされていません。 設立。.
免疫再構成症候群。
免疫再構成症候群が報告されています。 VIRACEPTを含む抗レトロウイルス療法の併用で治療された患者。中。 抗レトロウイルス療法の併用初期段階、その患者。 免疫系の反応は、怠惰または炎症反応を起こす可能性があります。 残存日和見感染症[Mycobacterium avium感染症など]。 サイトメガロウイルス、ニューモシスチスジロベチ肺炎(PCP)、または結核]。 さらなる評価と治療が必要になる場合があります。.
自己免疫疾患(グレイブス病など)。 多発性筋炎、ギランバレー症候群)も発生すると報告されています。 免疫再構成の設定;ただし、発症までの時間はもっと長くなります。 変動し、治療開始後何ヶ月も発生する可能性があります。.
患者カウンセリング情報。
見る。 FDA承認の患者。 ラベル付け(患者情報)。
患者への声明と。 医療提供者は、製品のボトルラベルに含まれています:ALERT:調べてください。 VIRACEPTと一緒に服用してはならない薬について
- 使用説明書。
最適な吸収のために。 患者は食物と一緒にVIRACEPTを服用するように助言されるべきです。.
患者さんには通知する必要があります。 VIRACEPTタブレットはフィルムコーティングされており、このフィルムコーティングは意図されています。 タブレットを飲み込みやすくします。.
VIRACEPTの用量の場合。 逃した、患者はできるだけ早く服用してから戻る必要があります。 彼らの通常のスケジュール。. ただし、用量をスキップした場合、患者はスキップしないでください。 次の線量を2倍にします。.
成人または小児患者。 錠剤を飲み込むことができないと、錠剤が少量溶解することがあります。 水:。
- VIRACEPTタブレットを配置します。 少量の水。
- 溶解したら、 ⁇ りを混ぜます。 よく液体し、すぐに消費します。.
- ガラスをすすいでください。 水とすすぎを飲み込んで、全量が確実に消費されるようにします。
できない小児患者。 ツバメ錠は粉末製剤を使用することもできます:。
- VIRACEPTオーラルパウダーをaと混合します。 少量の水、牛乳、調合乳、大豆粉ミルク、豆乳、または栄養補助食品。
- 混合すると、内容全体が表示されます。 全用量を得るために消費する必要があります。.
- 混合物が消費されない場合。 すぐに冷蔵保管する必要がありますが、保管を超えてはなりません。 6時間。.
- 酸性食品またはジュース(例:.、。 オレンジジュース、アップルジュース、アップルソース)は、混合にはお勧めしません。 組み合わせが苦い味をもたらす可能性があるため、VIRACEPTオーラルパウダー。.
- VIRACEPTオーラルパウダーはしないでください。 元の容器に水で再構成します。.
薬物相互作用。
VIRACEPTは一部と相互作用する可能性があります。 薬物;したがって、患者はその使用を医師に報告するように助言されるべきです。 その他の処方薬、非処方薬、ハーブ製品の。 特にセント. ジョンの麦 ⁇ 。.
経口投与されている患者。 避妊薬は、代替または追加の避妊薬として指示する必要があります。 VIRACEPTによる治療中は、対策を使用する必要があります。
シルデナフィルを投与されている患者。 または他のPDE5阻害剤、およびネルフィナビルは、それらが存在する可能性があることを通知する必要があります。 PDE5阻害剤関連の有害事象のリスク増加。 低血圧、視覚的変化、および長期の陰茎勃起、そして迅速にする必要があります。 症状があれば医師に報告してください。.
肝障害。
患者さんには通知する必要があります。 中等度または重度の肝がある場合は、VIRACEPTを使用しないでください。 障害。.
フェニルケトン尿症。
医師は警告する必要があります。 VIRACEPT経口粉末にフェニルアラニンが含まれているフェニルケトン尿症の患者。
脂肪の再分配。
患者さんには通知する必要があります。 体脂肪の再分布または蓄積は、投与中の患者で発生する可能性があること。 PREZISTA /リトナビルを含む抗レトロウイルス療法、およびその原因と。 これらの状態の長期的な健康への影響は現時点では不明です。
最も頻繁な有害事象。 VIRACEPTに関連するのは下 ⁇ で、通常は制御できます。 消化器を遅らせるロペラミドなどの非処方薬。 運動性。.
一般情報。
VIRACEPTは治療法ではありません。 HIV-1感染と患者は、関連する病気を経験し続ける可能性があります。 日和見感染症を含むHIV-1感染症。. 患者はすべきです。 VIRACEPTを使用するときは、医師の管理下に留まります。患者はすべきです。 HIV-1感染を他の人に広める可能性のあることを避けるようにアドバイスされました。.
- 針を共有しないでください。 その他の噴射装置。.
- 個人用品は共有しないでください。 歯ブラシやかみそりの ⁇ のように、血液や体液ができる。.
- なんのセックスもしないでください。 保護なし。. 常に安全に練習してください。 ラテックスまたはポリウレタンコンドームを使用して性的接触の可能性を下げることによるセックス。 精液、 ⁇ 分 ⁇ 物、または血液。.
- 母乳で育てないでください。. VIRACEPTを赤ちゃんに渡すことができるかどうかはわかりません。 あなたの母乳とそれがあなたの赤ちゃんに害を及ぼすかどうか。. また、HIV-1の母親。 HIV-1は乳房内の赤ちゃんに渡すことができるため、母乳を与えないでください。 牛乳。.
患者はそれを言われるべきです。 血漿HIV RNAの持続的な減少は、リスクの低下と関連しています。 エイズへの進行と死の。.
患者は下に留まるべきです。 VIRACEPTの使用中の医師のケア。患者さんには助言が必要です。 VIRACEPTと他の併用抗レトロウイルス療法を毎日服用してください。 処方された。. 患者は、相談なしに用量を変更したり、治療を中止したりしてはなりません。 彼らの医者と。.
非臨床毒性学。
発がん、変異誘発、生殖能力の障害。
マウスとラットの発がん性試験が行われた。 1000 mg / kg /日までの経口投与でネルフィナビルを使用。. 腫瘍形成の証拠はありません。 効果は、それらの9倍までの全身曝露(Cmax)でマウスで認められました。 ヒトで推奨される治療用量(750 mg TIDまたは1250 mg。 BID)。. ラットでは、甲状腺 ⁇ 胞細胞腺腫と癌腫が増加しました。 300 mg / kg /日以上の男性および1000 mg / kg /日の女性。. 全身。 300および1000 mg / kg /日の曝露(Cmax)は、それぞれ1〜3倍でした。 推奨される治療用量でヒトで測定されたもの。. 繰り返した。 ラットへのネルフィナビルの投与は、肝臓と一致する効果をもたらしました。 ミクロソーム酵素誘導と甲状腺ホルモン沈着の増加;これら。 甲状腺 ⁇ 胞細胞腫瘍に対するヒトではなく、素因ラットの影響。. ネルフィナビルは、バッテリーに変異原性または染色体異常誘発性の証拠を示さなかった。 の。 in vitro。 と。 in vivo。 遺伝毒物学アッセイ。. これらの研究が含まれています。 細菌変異アッセイ。 S.チフィムリウム。 と。 大腸菌。、マウス。 ヒトの染色体異常アッセイであるリンパ腫チロシンキナーゼアッセイ。 リンパ球、および。 in vivo。 マウスの骨髄小核アッセイ。.
ネルフィナビルは男性にも女性にも影響を与えませんでした。 全身曝露でのラットの交配と受胎能または胚生存は同等です。 人間の治療への曝露。.
特定の集団で使用します。
妊娠。
妊娠カテゴリーB
VIRACEPTは、妊娠中にのみ使用する必要があります。 潜在的な利益は、胎児への潜在的なリスクを正当化します。. ありません。 VIRACEPTを服用している妊婦を対象とした適切で適切に管理された研究。
胎児の発育や母親への影響はありませんでした。 全身曝露で妊娠中のラットにネルフィナビルを投与した場合の毒性。 (AUC)人間の曝露に匹敵します。. 妊娠へのネルフィナビルの投与。 ウサギは、わずかな用量まで胎児の発育効果をもたらさなかった。 母体体重の減少が観察された。ただし、最高でも。 用量評価では、ウサギの全身曝露はヒトよりも有意に低かった。 露出。. ラットを用いた追加の研究は、ネルフィナビルへの暴露が 妊娠中から授乳までの女性は生存に影響を与えませんでした。 成長、そして離乳への子孫の発達。. その後の生殖。 これらの子孫のパフォーマンスも、母体への曝露の影響を受けませんでした。 ネルフィナビル。.
抗レトロウイルス妊娠登録(APR)。
妊婦の母胎児転帰を監視する。 VIRACEPTおよびその他の抗レトロウイルス薬、抗レトロウイルス薬に曝露。 妊娠登録が確立されました。. 医師は登録することをお勧めします。 (800)258-4263に電話して患者。.
授乳中の母親。
疾病管理予防センター。 HIVに感染した母親は、リスクを回避するために乳児に母乳を与えないことを推奨します。 HIVの出生後感染 授乳中のラットを用いた研究が実証されています。 そのネルフィナビルは牛乳中に排 ⁇ されます。. HIVの可能性の両方のため。 乳児における感染と深刻な副作用の可能性。 母親は、VIRACEPTを受けている場合は母乳を与えないように指示する必要があります。
小児用。
安全性、忍容性、薬物動態プロファイルおよび。 VIRACEPTの有効性は、HIVに感染した小児患者で2からまで評価されました。 多施設臨床試験、研究556およびPACTG 337の13歳。. 2年未満の患者。 年齢、VIRACEPTは研究された用量で安全であることがわかりましたが、確実に。 有効量を確立できませんでした。. 薬物動態プロファイル、。 青年期の患者におけるVIRACEPTの安全性と抗ウイルス活性13年。 古いものは、いくつかの試験がある成人の臨床試験のデータによってサポートされています。 13歳以上の被験者の登録を許可しました。. したがって、のデータ。 青年および成人は集合的に分析された。.
老人用。
VIRACEPTの臨床試験には十分ではありませんでした。 65歳以上の被験者の数で、反応が異なるかどうかを判断します。 若い科目から。.
肝障害。
VIRACEPTは、どちらかの患者には使用しないでください。 中等度または重度の肝障害(Child-Pugh BまたはC、スコアがまたはより大きい。 7に等しい)。. 軽度の肝患者の場合、VIRACEPTの用量調整は必要ありません。 障害(Child-Pugh A、スコア5-6)。.
腎障害。
VIRACEPTの安全性と有効性はそうではありません。 腎障害のある患者で確立されました。.
CYP3A and CYP2C19 appear to be the predominant enzymes that metabolize nelfinavir in humans. The potential ability of nelfinavir to inhibit the major human cytochrome P450 enzymes (CYP3A, CYP2C19, CYP2D6, CYP2C9, CYP1A2 and CYP2E1) has been investigated in vitro. Only CYP3A was inhibited at concentrations in the therapeutic range. Specific drug interaction studies were performed with nelfinavir and a number of drugs. Table 12 summarizes the effects of nelfinavir on the geometric mean AUC, Cmax and Cmin of coadministered drugs. Table 13 shows the effects of coadministered drugs on the geometric mean AUC, Cmax and Cmin of nelfinavir.
Table 12: Drug Interactions: Changes in Pharmacokinetic Parameters for Coadministered Drug in the Presence of VIRACEPT
| Coadministered Drug | Nelfinavir Dose | N | % Change of Coadministered Drug Pharmacokinetic Parameters* (90% CI) | ||
| AUC | Cmax | Cmin | |||
| HIV-Protease Inhibitors | |||||
| Indinavir 800 mg Single Dose | 750 mg q8h x 7 days | 6 | ↑51% (↑29-↑77%) |
↓10% (↓28-↑13%) |
NA |
| Ritonavir 500 mg Single Dose | 750 mg q8h x 5 doses | 10 | ↔ | ↔ | NA |
| Saquinavir 1200 mg Single Dose† | 750 mg TID x 4 days | 14 | ↑392% (↑291-↑521%) |
↑179% (↑117-↑259%) |
NA |
| Amprenavir 800 mg TID x 14 days | 750 mg TID x 14 days | 6 | ↔ | ↓14% (↓38-↑20%) |
↑189% (↑52-↑448%) |
| Nucleoside Reverse Transcriptase Inhibitors | |||||
| Lamivudine 150 mg Single Dose | 750 mg q8h x 7-10 days | 11 | ↑10% (↑2-↑18%) |
↑31% (↑9-↑56%) |
NA |
| Zidovudine 200 mg Single Dose | 750 mg q8h x 7-10 days | 11 | ↓35% (↓29-↓40%) |
↓31% (↓13-↓46%) |
NA |
| Non-nucleoside Reverse Transcriptase Inhibitors | |||||
| Efavirenz 600 mg qd x 7 days | 750 mg q8h x 7 days | 10 | ↓12% (↓31-↑12%) |
↓12% (↓29-↑8%) |
↓22% (↓54-↑32%) |
| Delavirdine 400 mg q8h x 14 days | 750 mg q8h x 7 days | 7 | ↓31% (↓57-↑10%) |
↓27% (↓49-↑4%) |
↓33% (↓70-↑49%) |
| Anti-infective Agents | |||||
| Rifabutin 150 mg qd x 8 days§ | 750 mg q8h x 7-8 days1 | 12 | ↑83% (↑72-↑96%) |
↑19% (↑11-↑28%) |
↑177% (↑144-↑215%) |
| Rifabutin 300 mg qd x 8 days | 750 mg q8h x 7-8 days | 10 | ↑207% (↑161-↑263%) |
↑146% (↑118-↑178%) |
↑305% (↑245-↑375%) |
| Azithromycin 1200 mg Single Dose | 750 mg TID x 11 days | 12 | ↑112% (↑80-↑150%) |
↑136% (↑77-↑215%) |
NA |
| HMG-CoA Reductase Inhibitors | |||||
| Atorvastatin 10 mg qd x 28 days | 1250 mg BID x 14 days | 15 | ↑74% (↑41-↑116%) |
↑122% (↑68-↑193%) |
↑39% (↓21-↑145%) |
| Simvastatin 20 mg qd x 28 days | 1250 mg BID x 14 days | 16 | ↑505% (↑393-↑643%) |
↑517% (↑367-↑715%) |
ND |
| Other Agents | |||||
| Ethinyl estradiol 35 |ag qd x 15 days | 750 mg q8h x 7 days | 12 | ↓47% (↓42-↓52%) |
↓28% (↓16-↓37%) |
↓62% (↓57-↓67%) |
| Norethindrone 0.4 mg qd x 15 days | 750 mg q8h x 7 days | 12 | ↓18% (↓13-↓23%) |
↔ | ↓46% (↓38-↓53%) |
| Methadone 80 mg ± 21 mg qd# > 1 month | 1250 mg BID x 8 days | 13 | ↓47% (↓42-↓51%) |
↓46% (↓42-↓49%) |
↓53% (↓49-↓57%) |
| Phenytoin 300 mg qd x 14 daysÞ | 1250 mg BID x 7 days | 12 | ↓29% (↓17-↓39%) |
↓21% (↓12-↓29%) |
↓39% (↓27-↓49%) |
| NA: Not relevant for single-dose treatment; ND: Cannot be
determined * ↑ Indicates increase; ↓ Indicates decrease; ↔ Indicates no change (geometric mean exposure increased, or decreased < 10%) † Using the soft-gelatin capsule formulation of saquinavir 1200 mg § Rifabutin 150 mg qd changes are relative to Rifabutin 300 mg qd x 8 days without coadministration with nelfinavir ¶Comparable changes in rifabutin concentrations were observed with VIRACEPT 1250 mg q12h x 7 days # Changes are reported for total plasma methadone; changes for the individual R-enantiomer and S-enantiomer were similar Þ Phenytoin exposure measures are reported for total phenytoin exposure. The effect of nelfinavir on unbound phenytoin was similar |
|||||
Table 13: Drug Interactions: Changes in
Pharmacokinetic Parameters for Nelfinavir in the Presence of the Coadministered
Drug
| Coadministered Drug | Nelfinavir Dose | N | % Change of Nelfinavir Pharmacokinetic Parameters* (90% CI) | ||
| AUC | C max | C min | |||
| HIV-Protease Inhibitors | |||||
| Indinavir 800 mg q8h x 7 days | 750 mg Single Dose | 6 | ↑83% (↑42-↑137%) |
↑31% (↑16-↑48%) |
NA |
| Ritonavir 500 mg q12h x 3 doses | 750 mg Single Dose | 10 | ↑152% (↑96-↑224%) |
↑44% (↑28-↑63%) |
NA |
| Saquinavir 1200 mg TID x 4 dayst | 750 mg Single Dose | 14 | ↑18% (↑7-↑30%) |
↔ | NA |
| Nucleoside Reverse Transcriptase Inhibitors | |||||
| Didanosine 200 mg Single Dose | 750 mg Single Dose | 9 | ↔ | ↔ | NA |
| Zidovudine 200 mg + Lamivudine 150 mg Single Dose | 750 mg q8h x 7-10 days | 11 | ↔ | ↔ | ↔ |
| Non-nucleoside Reverse Transcriptase Inhibitors | |||||
| Efavirenz 600 mg qd x 7 days | 750 mg q8h x 7 days | 7 | ↑20% (↑8-↑34%) |
↑21% (↑10-↑33%) |
↔ |
| Nevirapine 200 mg qd x 14 days followed by 200 mg BID x 14 days | 750 mg TID x 36 days | 23 | ↔ | ↔ | ↓32% (↓50-↑5%) |
| Delavirdine 400 mg q8h x 7 days | 750 mg q8h x 14 days | 12 | ↑107% (↑83-↑135%) |
↑88% (↑66-↑113%) |
↑136% (↑103-↑175%) |
| Anti-infective Agents | |||||
| Ketoconazole 400 mg qd x 7 days | 500 mg q8h x 5-6 days | 12 | ↑35% (↑24-↑46%) |
↑25% (↑11-↑40%) |
↑14% (↓23-↑69%) |
| Rifabutin 150 mg qd x 8 days | 750 mg q8h x 7-8 days | 11 | ↓23% (↓14-↓31%) |
↓18% (↓8-↓27%) |
↓25% (↓8-↓39%) |
| 1250 mg q12h x 7-8 days | 11 | ↔ | ↔ | ↓15% (↓43-↑27%) |
|
| Rifabutin 300 mg qd x 8 days | 750 mg q8h x 7-8 days | 10 | ↓32% (↓15-↓46%) |
↓24% (↓10-↓36%) |
↓53% (↓15-↓73%) |
| Rifampin 600 mg qd x 7 days | 750 mg q8h x 5-6 days | 12 | ↓83% (↓79-↓86%) |
↓76% (↓69-↓82%) |
↓92% (↓86-↓95%) |
| Azithromycin 1200 mg Single Dose | 750 mg tid x 9 days | 12 | ↓15% (↓7-↓22%) |
↓10% (↓19-↑1%) |
↓29% (↓19-↓38%) |
| Other Agents | |||||
| Phenytoin 300 mg qd x 7 days | 1250 mg BID x 14 days | 15 | ↔ | ↔ | ↓18% (↓45-↑23%) |
| Omeprazole 40 mg qd x 4 days administered 30 minutes before nelfinavir | 1250 mg BID x 4 days | 19 | ↓36% (↓20-↓49%) |
↓37% (↓23-↓49%) |
↓39% (↓15-↓57%) |
| NA: Not relevant for single-dose treatment * ↑ Indicates increase; ↓ Indicates decrease; ↔ Indicates no change (geometric mean exposure increased or decreased < 10%) † Using the soft-gelatin capsule formulation of saquinavir 1200 mg |
|||||
妊娠カテゴリーB
VIRACEPTは、妊娠中にのみ使用する必要があります。 潜在的な利益は、胎児への潜在的なリスクを正当化します。. ありません。 VIRACEPTを服用している妊婦を対象とした適切で適切に管理された研究。
胎児の発育や母親への影響はありませんでした。 全身曝露で妊娠中のラットにネルフィナビルを投与した場合の毒性。 (AUC)人間の曝露に匹敵します。. 妊娠へのネルフィナビルの投与。 ウサギは、わずかな用量まで胎児の発育効果をもたらさなかった。 母体体重の減少が観察された。ただし、最高でも。 用量評価では、ウサギの全身曝露はヒトよりも有意に低かった。 露出。. ラットを用いた追加の研究は、ネルフィナビルへの暴露が 妊娠中から授乳までの女性は生存に影響を与えませんでした。 成長、そして離乳への子孫の発達。. その後の生殖。 これらの子孫のパフォーマンスも、母体への曝露の影響を受けませんでした。 ネルフィナビル。.
抗レトロウイルス妊娠登録(APR)。
妊婦の母胎児転帰を監視する。 VIRACEPTおよびその他の抗レトロウイルス薬、抗レトロウイルス薬に曝露。 妊娠登録が確立されました。. 医師は登録することをお勧めします。 (800)258-4263に電話して患者。.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Experience: Adults And Adolescents (13 Years and Older)
The safety of VIRACEPT was studied in over 5000 patients who received drug either alone or in combination with nucleoside analogues. The majority of adverse events were of mild intensity. The most frequently reported adverse event among patients receiving VIRACEPT was diarrhea, which was generally of mild to moderate intensity.
Drug-related clinical adverse experiences of moderate or severe intensity in ≥ 2% of patients treated with VIRACEPT coadministered with d4T and 3TC (Study 542) for up to 48 weeks, or with ZDV plus 3TC (Study 511) for up to 24 weeks are presented in Table 4.
Table 4: Percentage of Patients with
Treatment-Emergent* Adverse Events of Moderate or Severe Intensity Reported in ≥
2% of Adult and Adolescent Patients
| Adverse Events | Study 511 24 weeks | Study 542 48 weeks | |||
| Placebo + ZDV/3TC (n=101) |
500 mg TID VIRACEPT + ZDV/3TC (n=97) |
750 mg TID VIRACEPT + ZDV/3TC (n=100) |
1250 mg BID VIRACEPT + d4T/3TC (n=344) |
750 mg TID VIRACEPT + d4T/3TC (n=210) |
|
| Digestive System | |||||
| Diarrhea | 3% | 14% | 20% | 20% | 15% |
| Nausea | 4% | 3% | 7% | 3% | 3% |
| Flatulence | 0 | 5% | 2% | 1% | 1% |
| Skin/Appendages | |||||
| Rash | 1% | 1% | 3% | 2% | 1% |
| * Includes those adverse events at least possibly, probably or definitely related to study drug or of unknown relationship and excludes concurrent HIV conditions | |||||
Adverse events occurring in less than 2% of patients receiving VIRACEPT in all phase 2 and 3 clinical trials and considered at least possibly related or of unknown relationship to treatment and of at least moderate severity are listed below.
Body as a Whole: abdominal pain, accidental injury, allergic reaction, asthenia, back pain, fever, headache, malaise, pain, and redistribution/accumulation of body fat.
Digestive System: anorexia, dyspepsia, epigastric pain, gastrointestinal bleeding, hepatitis, mouth ulceration, pancreatitis, and vomiting.
Hemic/Lymphatic System: anemia, leukopenia, and thrombocytopenia.
Metabolic/Nutritional System: increases in alkaline phosphatase, amylase, creatine phosphokinase, lactic dehydrogenase, SGOT, SGPT, and gamma-glutamyl transpeptidase; hyperlipemia, hyperuricemia, hyperglycemia, hypoglycemia, dehydration, and liver function tests abnormal.
Musculoskeletal System: arthralgia, arthritis, cramps, myalgia, myasthenia, and myopathy.
Nervous System: anxiety, depression, dizziness, emotional lability, hyperkinesia, insomnia, migraine, paresthesia, seizures, sleep disorder, somnolence, and suicide ideation.
Respiratory System: dyspnea, pharyngitis, rhinitis, and sinusitis.
Skin/Appendages: dermatitis, folliculitis, fungal dermatitis, maculopapular rash, pruritus, sweating, and urticaria.
Special Senses: acute iritis and eye disorder.
Urogenital System: kidney calculus, sexual dysfunction, and urine abnormality.
Laboratory Abnormalities
The percentage of patients with marked laboratory abnormalities in Studies 542 and 511 are presented in Table 5. Marked laboratory abnormalities are defined as a Grade 3 or 4 abnormality in a patient with a normal baseline value, or a Grade 4 abnormality in a patient with a Grade 1 abnormality at baseline.
Table 5: Percentage of Patients by Treatment Group
with Marked Laboratory Abnormalities* in > 2% of Patients
| Study 511 | Study 542 | ||||
| Placebo+ ZDV/3TC (n=101) |
500 mg TID VIRACEPT +ZDV/3TC (n=97) |
750 mg TID VIRACEPT +ZDV/3TC (n=100) |
1250 mg BID VIRACEPT+ d4T/3TC (n=344) |
750 mg TID VIRACEPT+ d4T/3TC (n=210) |
|
| Hematology | |||||
| Hemoglobin | 6% | 3% | 2% | 0 | 0 |
| Neutrophils | 4% | 3% | 5% | 2% | 1% |
| Lymphocytes | 1% | 6% | 1% | 1% | 0 |
| Chemistry | |||||
| ALT (SGPT) | 6% | 1% | 1% | 2% | 1% |
| AST (SGOT) | 4% | 1% | 0 | 2% | 1% |
| Creatine Kinase | 7% | 2% | 2% | NA | NA |
| * Marked laboratory abnormalities are defined as a shift from Grade 0 at baseline to at least Grade 3 or from Grade 1 to Grade 4 | |||||
Clinical Trials Experience: Pediatrics (2 to Less than 13 Years of Age)
VIRACEPT has been studied in approximately 400 pediatric patients in clinical trials from birth to 13 years of age. The adverse event profile seen during pediatric clinical trials was similar to that for adults.
The most commonly reported drug-related, treatment-emergent adverse events reported in the pediatric studies included: diarrhea, leukopenia/neutropenia, rash, anorexia, and abdominal pain. Diarrhea, regardless of assigned relationship to study drug, was reported in 39% to 47% of pediatric patients receiving VIRACEPT in 2 of the larger treatment trials. Leukopenia/neutropenia was the laboratory abnormality most commonly reported as a significant event across the pediatric studies.
Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VIRACEPT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: hypersensitivity reactions (including bronchospasm, moderate to severe rash, fever, and edema).
Cardiovascular System: QTc prolongation, torsades de pointes.
Digestive System: jaundice.
Metabolic/Nutritional System: bilirubinemia, metabolic acidosis.
VIRACEPTによる急性過剰摂取の人間の経験は、 限定。. VIRACEPTの過剰摂取に対する特定の解毒剤はありません。示されている場合、。 吸収されない薬物の排除は、 ⁇ 吐または胃洗浄によって達成されるべきである。. 活性炭の投与は、除去を助けるためにも使用されるかもしれません。 吸収されない薬物。. ネルフィナビルはタンパク質に高度に結合しているため、透析は起こりそうにありません。 血液から薬物を大幅に除去する。.
Effects on Electrocardiogram
The effect of Viracept at the recommended dose of 1250 mg twice daily on the QTcF interval administered with a low fat meal (20% fat) was evaluated in a randomized, placebo and active (moxifloxacin 400 mg once daily) controlled, crossover study in 66 healthy subjects. The maximum mean time-matched (95% upper confidence bound) differences in QTcF interval from placebo after baselinecorrection was below 10 milliseconds, the threshold of clinical concern. This finding was unchanged when a single supratherapeutic dose of Viracept 3125 mg was administered following a 3-day administration of Viracept 1250 mg twice daily. The exposure at 3125 mg was 1.4-fold that at 1250 mg. The dose of 3125 mg in this study did not achieve the anticipated exposures in patients taking a high fat meal (50% fat) or with concomitant administration of drugs that could increase nelfinavir exposure.
No subject in any group had an increase in QTcF of ≥ 60 milliseconds from baseline. No subject experienced an interval exceeding the potentially clinically relevant threshold of 500 milliseconds.
ネルフィナビルの薬物動態学的特性はあった。 健康なボランティアとHIV感染患者で評価。実質的な違いはありません。 2つのグループ間で観察されました。.
吸収。
ネルフィナビルの薬物動態パラメーター(下の領域。 定常状態での24時間の血漿濃度-時間曲線[AUC24]、。 ピーク血漿濃度[Cmax]、朝と夕方のトラフ濃度。 [Ctrough])複数後のHIV陽性患者を対象とした薬物動態研究から。 1250 mg(5つの250 mg錠剤)を1日2回(BID)28日間(10。 患者)および750 mg(3つの250 mgタブレット)を1日3回(TID)28日間。 (11人の患者)を表7にまとめます。.
表7:薬物動態研究の要約。
1250 mg(5 250 mg錠剤)BIDの複数回投与のHIV陽性患者。
28日間および750 mg(3つの250 mgタブレット)TIDで28日間。
| レジメン。 | AUC24。 mg•h / L。 |
Cmax。 mg / L。 |
Ctrough。 朝。 mg / L。 |
Ctrough。 午後か夕方。 mg / L。 |
| 1250 mg BID。 | 52.8±15.7。 | 4.0±0.8。 | 2.2±1.3。 | 0.7±0.4。 |
| 750 mg TID。 | 43.6±17.8。 | 3.0±1.6。 | 1.4±0.6。 | 1.0±0.5。 |
| データは平均±SDです。 |
朝と午後または夕方の違い。 TIDおよびBIDレジメンのトラフ濃度も健康で観察されました。 正確に8時間または12時間間隔で投与されたボランティア。.
1250 mgの単回投与を受けている健康なボランティアでは、 625 mgタブレットは、250 mgタブレット製剤と生物学的に同等ではありませんでした。. 下。 空腹時条件(n = 27)、AUCとCmaxは34%と24%高くなりました。 それぞれ、625 mg錠。. 相対的なバイオアベイラビリティ評価。 摂食条件下(n = 28)では、625 mg錠剤のAUCは24%高くなりました。 。 Cmaxは両方の製剤で同等でした。. HIV-1感染被験者(N = 21)。 摂食条件下で1250 mg BIDを複数回投与する625 mg製剤。 定常状態の類似性に基づく250 mg製剤と生物学的に同等でした。 ばく露(CmaxおよびAUC)。.
表8は、定常状態の要約を示しています。 複数回投与後のネルフィナビルの薬物動態パラメーター(平均±SD)。 HIV感染患者(N = 21)に対する1250 mg BID(2 x 625 mg錠)の14。 日。.
表8:定常状態の薬物動態の概要。
1250の複数回投与後のネルフィナビルのパラメーター(平均±SD)。
14日間のHIV感染患者(N = 21)へのBID mg(2 x 625 mg錠剤)。.
| レジメン。 | AUC12。 mg•h / L。 |
Cmax。 mg / L。 |
Cmin。 mg / L。 |
| 1250 mg BID。 | 35.3(16.4)。 | 4.7(1.9)。 | 1.5(1.0)。 |
| AUC12:定常状態AUC。 Cmax:定常状態での最大血漿濃度。 Cmin:定常状態での最小血漿濃度。 |
健康なボランティアでは、750 mgを1回投与します。 摂食条件下では、ネルフィナビル濃度は投与後も同様でした。 250 mgの錠剤と経口粉末の。.
経口吸収に対する食物の影響。
食物はネルフィナビルへの曝露を増加させ、減少させます。 空腹状態に対するネルフィナビルの薬物動態学的変動。. 一つに。 研究では、健康なボランティアが1250 mgのVIRACEPT 250 mgの単回投与を受けました。 空腹時または摂食時の錠剤(5錠)(3種類の食事)。. に。 2番目の研究では、健康なボランティアが1250 mg VIRACEPTの単回投与を受けました(5。 x 250 mg錠)空腹時または摂食時(2つの異なる脂肪含有量)。 食事)。. 2つの研究の結果を表9と表にまとめます。 それぞれ10。.
表9:ネルフィナビルのAUC、Cmax、Tmaxの増加。
1250 mg VIRACEPT(5 x 250 mg。
タブレット)。
| Kcalの数。 | %脂肪。 | 被験者の数。 | AUCの折り目が増加します。 | Cmax折り目増加。 | Tmax(hr)の増加。 |
| 125。 | 20 | n = 21。 | 2.2。 | 2.0。 | 1.00。 |
| 500。 | 20 | n = 22。 | 3.1。 | 2.3。 | 2.00。 |
| 1000。 | 50 | n = 23。 | 5.2。 | 3.3。 | 2.00。 |
表10:ネルフィナビルAUC、Cmax、Tmaxの増加。
Fed低脂肪(20%)対高脂肪(50%)の状態は、破 ⁇ 状態に関連しています。
1250 mg VIRACEPT(5 x 250 mgタブレット)に続いて。
| Kcalの数。 | %脂肪。 | 被験者の数。 | AUCの折り目が増加します。 | Cmax折り目増加。 | Tmax(hr)の増加。 |
| 500。 | 20 | n = 22。 | 3.1。 | 2.5。 | 1.8。 |
| 500。 | 50 | n = 22。 | 5.1。 | 3.8。 | 2.1。 |
ネルフィナビルへの曝露は、 VIRACEPTで摂取した食事のカロリーまたは脂肪含有量。
625では、食品効果の研究は行われていません。 mgタブレット。. ただし、クロススタディの比較に基づく(n = 26フィードvs. n = 26断食)。 ネルフィナビル1250 mgの単回投与後、 625 mgのネルフィナビル錠剤の食物効果は、それに匹敵するようです。 250 mgの錠剤。. VIRACEPTは食事と一緒に服用する必要があります。.
分布。
経口後の分布の見かけの量。 ネルフィナビルの投与は2-7 L / kgでした。. 血清中のネルフィナビルは広範囲にタンパク質結合しています。 (> 98%)。.
代謝。
変化のないネルフィナビルは総血漿の82-86%を占めていました。 14C-ネルフィナビルの単回経口750 mg投与後の放射能。. In vitro。、 複数。 CYP3AおよびCYP2C19を含むチトクロームP-450酵素が原因です。 ネルフィナビルの代謝。. 1つの主要なおよびいくつかの小さな酸化代謝物。 血漿中に発見されました。. 主要な酸化代謝物があります。 in vitro。 抗ウイルス。 親薬に匹敵する活動。.
除去。
血漿中の最終的な半減期は通常3.5から5でした。 時間。. 14C-ネルフィナビルを含む経口750 mg用量の大部分(87%)が回収されました。 ⁇ 便中; ⁇ 便の放射能は多数の酸化的代謝物で構成されていました。 (78%)と変化のないネルフィナビル(22%)。. 用量の1-2%だけが回収されました。 尿、変化のないネルフィナビルが主要な成分でした。.

































