

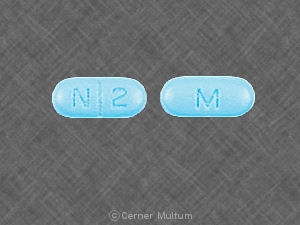
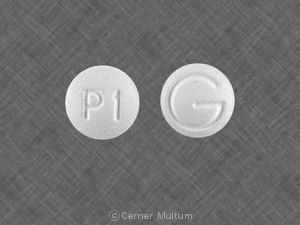
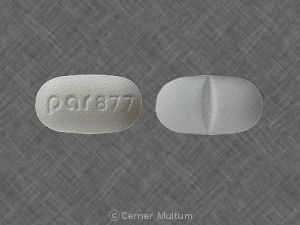
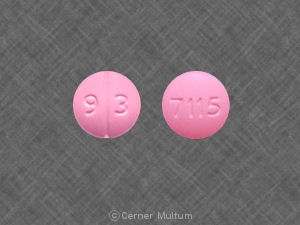
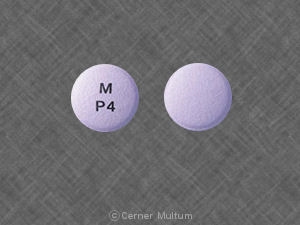
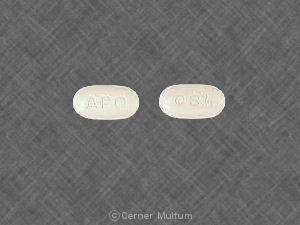

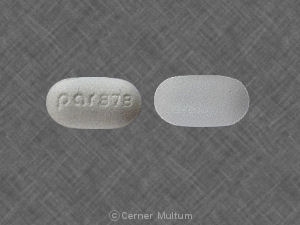
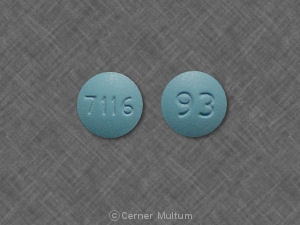








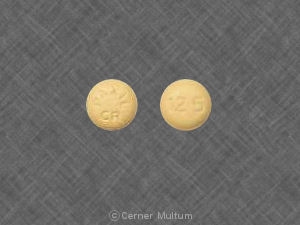



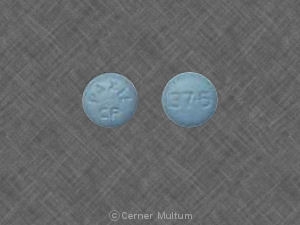







Composition:
Application:
Utilisé dans le traitement:
Examiné médicalement par Militian Inessa Mesropovna, Pharmacie Dernière mise à jour le 26.06.2023

Attention! Information sur la page est réservée aux professionnels de la santé! Les informations sont collectées dans des sources ouvertes et peuvent contenir des erreurs significatives! Soyez prudent et revérifiez toutes les informations de cette page!
Comprimés
Comprimés pelliculés, ovales modifiés, comme suit:
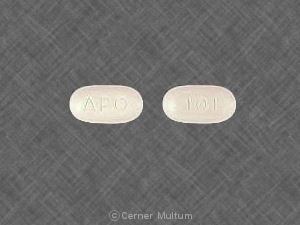
10 mg comprimés blancs avec l'inscription POT 10 sur un côté. NDC 54766-201-01 Bouteilles de 30
20 mg comprimés orange foncé avec l'inscription POT 20 allumée d'un côté. Les comprimés sont sécables des deux côtés. NDC 54766-202-01 Bouteilles de 30
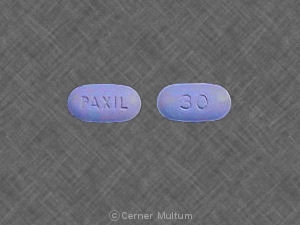
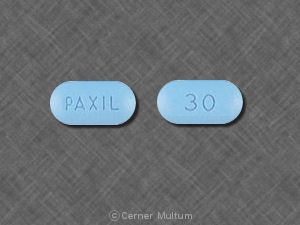
30 mg comprimés jaunes avec l'inscription POT 30 sur un côté. NDC 54766-203-01 Bouteilles de 30
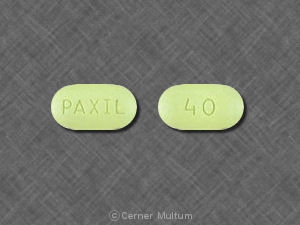
40 mg comprimés de rose avec l'inscription POT 40 sur une face. NDC 54766-204-01 Bouteilles de 30
Protéger de l'humidité. Conserver à 25 ° C (77 ° F) ; excursions autorisées à 15 ° -30 ° C (59 ° et 86 ° F) (voir Salle contrôlée USP Température)
Distribué par Sebela Pharmaceuticals Inc. 645 Hembree Parkway, Suite I Roswell, Géorgie 30076. www.sebelapharma.com. Sans frais 1-844-732-3521. Révisé: Jan 2017
Trouble dépressif majeur
PEXEVA® (mesylate de paroxétine) est indiqué pour le traitement du MDD .
L'efficacité de la paroxétine dans le traitement d'une majeure un épisode dépressif a été établi dans des essais contrôlés de 6 semaines sur des patients externes dont les diagnostics correspondaient le plus étroitement à la catégorie DSM-III de MDD (voir PHARMACOLOGIE CLINIQUE). Un épisode dépressif majeur implique un proéminent et humeur déprimée ou dysphorique relativement persistante qui interfère généralement avec fonctionnement quotidien (presque tous les jours pendant au moins 2 semaines); il doit inclure à au moins 4 des 8 symptômes suivants: changement d'appétit, changement de sommeil agitation ou retard psychomoteur, perte d'intérêt pour les activités habituelles ou diminution de la libido, augmentation de la fatigue, sentiments de culpabilité ou impuissance, ralentissement de la réflexion ou altération de la concentration, et tentative de suicide ou idées suicidaires.
Les effets de la paroxétine chez les personnes hospitalisées déprimées les patients n'ont pas été suffisamment étudiés.
L'efficacité de la paroxétine dans le maintien d'une réponse Le MDD pendant une période pouvant aller jusqu'à 1 an a été démontré dans un essai contrôlé par placebo (voir PHARMACOLOGIE CLINIQUE). Néanmoins, le médecin qui choisit d'utiliser PEXEVA® (mesylate de paroxétine) pendant de longues périodes devrait périodiquement réévaluer l'utilité à long terme du médicament pour chaque patient.
Trouble obsessionnel compulsif
PEXEVA® (mesylate de paroxétine) est indiqué pour le traitement des obsessions et des compulsions chez les patients atteints de TOC tel que défini dans le DSM-IV. Les obsessions ou compulsions provoquent une détresse marquée, sont long ou gênant de manière significative les relations sociales ou professionnelles fonctionnement.
L'efficacité de la paroxétine a été établie en deux semaines essais avec des patients externes obsessionnels compulsifs dont les diagnostics correspondaient le plus étroitement à la catégorie DSM-IIIR des TOC (voir Clinique Essais).
Le TOC se caractérise par des idées récurrentes et persistantes pensées, impulsions ou images (obsessions) qui sont égodystoniques et / ou les comportements répétitifs, délibérés et intentionnels (compulsions) qui le sont reconnu par la personne comme excessif ou déraisonnable.
Le maintien à long terme de l'efficacité a été démontré dans un Essai de prévention des rechutes de 6 mois. Dans cet essai, les patients assignés à la paroxétine a montré un taux de rechute inférieur à celui des patients sous placebo (voir Essais cliniques). Néanmoins, le médecin qui choisit d'utiliser PEXEVA® (mésylate de paroxétine) pendant de longues périodes devrait réévaluer périodiquement l'utilité à long terme du médicament pour l'individu patient (voir DOSAGE ET ADMINISTRATION).
Trouble panique
PEXEVA® (mesylate de paroxétine) est indiqué pour le traitement de la PD, avec ou sans agoraphobie, tel que défini dans DSM-IV. PD est caractérisé par la survenue d'attaques de panique inattendues et associées inquiétude d'avoir des attaques supplémentaires, s'inquiéter des implications ou conséquences des attaques et / ou un changement significatif de comportement lié à les attaques.
L'efficacité de la paroxétine a été établie en trois 10 à Essais de 12 semaines chez des patients atteints de DP dont les diagnostics correspondaient au DSM-IIIR catégorie de PD (voir Essais cliniques).
PD (DSM-IV) se caractérise par un imprévu récurrent attaques de panique, c'est-à-dire une période discrète de peur intense ou d'inconfort dans laquelle 4 (ou plus) des symptômes suivants se développent brusquement et atteignent un pic à moins de 10 minutes: (1) palpitations, battement de cœur ou accélération du rythme cardiaque; (2) transpiration; (3) tremblant ou tremblant; (4) sensations d'essoufflement ou étouffement; (5) sensation d'étouffement; (6) douleur thoracique ou inconfort; (7) nausées ou détresse abdominale; (8) sensation de vertige, instable, étourdi ou faible; (9) déréalisation (sentiments d'irréalité) ou dépersonnalisation (détaché de soi-même); (10) peur de perdre le contrôle; (11) peur de mourir; (12) paresthésies (sensations d'engourdissement ou de picotements); (13) frissons ou bouffées de chaleur.
Le maintien à long terme de l'efficacité a été démontré dans un Essai de prévention des rechutes de 3 mois. Dans cet essai, les patients atteints de DP sont affectés à la paroxétine a démontré un taux de rechute inférieur à celui des patients sous placebo (voir Essais cliniques). Néanmoins, le médecin qui prescrit PEXEVA® (mesylate de paroxétine) pendant de longues périodes devrait réévaluer périodiquement l'utilité à long terme du médicament pour le patient individuel.
Trouble d'anxiété généralisée
La paroxétine est indiquée pour le traitement de la généralisation Trouble anxieux (TAG), tel que défini dans DSM-IV. Anxiété ou tension associée avec le stress de la vie quotidienne ne nécessite généralement pas de traitement avec un anxiolytique.
L'efficacité de la paroxétine dans le traitement du GAD était établi dans deux essais contrôlés contre placebo de 8 semaines chez des adultes atteints de TAG La paroxétine n'a pas été étudiée chez les enfants ou les adolescents généralisés Trouble anxieux (voir Essais cliniques).
Le trouble d'anxiété généralisée (DSM-IV) est caractérisé par anxiété et inquiétude excessives (attente inquiétante) persistantes au moins 6 mois et que la personne a du mal à contrôler. Ça doit être associé à au moins 3 des 6 symptômes suivants: agitation ou sensation clé vers le haut ou sur le bord, étant facilement fatiguée, difficulté à se concentrer ou à l'esprit devenir vide, irritabilité, tension musculaire, troubles du sommeil.
L'efficacité de la paroxétine dans le maintien d'une réponse patients atteints d'un trouble d'anxiété généralisée, qui ont répondu pendant 8 semaines phase de traitement aigu lors de la prise de paroxétine et ont ensuite été observés rechute pendant une période pouvant aller jusqu'à 24 semaines, a été démontrée dans un essai contrôlé par placebo (voir Essais cliniques). Néanmoins, le médecin qui choisit d'utiliser la paroxétine pendant de longues périodes devrait réévaluer périodiquement l'utilité à long terme du médicament pour le patient individuel (voir DOSAGE ET ADMINISTRATION).
Trouble dépressif majeur
Posologie initiale habituelle
PEXEVA® (mesylate de paroxétine) doit être administré en tant que a dose quotidienne unique avec ou sans nourriture, généralement le matin. Le recommandé la dose initiale est de 20 mg / jour. Les patients ont été dosés dans une plage de 20 à 50 mg / jour les essais cliniques démontrant l'efficacité de la paroxétine dans le traitement du MDD. Comme pour tous les médicaments efficaces dans le traitement du MDD, le plein l'effet peut être retardé. Certains patients ne répondant pas à une dose de 20 mg peuvent en bénéficier des augmentations de dose, par incréments de 10 mg / jour, jusqu'à un maximum de 50 mg / jour. Les changements de dose doivent se produire à des intervalles d'au moins 1 semaine.
Thérapie d'entretien
Il n'y a pas de preuves disponibles pour répondre à la question de la durée pendant laquelle le patient traité par la paroxétine doit y rester. Il est généralement admis que les épisodes aigus de MDD nécessitent plusieurs mois ou plus long d'un traitement pharmacologique soutenu. Si la dose devait induire la rémission est identique à la dose nécessaire pour maintenir et / ou maintenir l'euthymie est inconnu.
Évaluation systématique de l'efficacité de la paroxétine a a montré que l'efficacité est maintenue pendant des périodes allant jusqu'à 1 an avec des doses qui en moyenne environ 30 mg.
Trouble obsessionnel compulsif
Posologie initiale habituelle
PEXEVA® (mesylate de paroxétine) doit être administré en tant que a dose quotidienne unique avec ou sans nourriture, généralement le matin. Le recommandé la dose de paroxétine dans le traitement de l'OCD est de 40 mg par jour. Les patients devraient l'être a commencé à 20 mg / jour et la dose peut être augmentée par incréments de 10 mg / jour. Les changements de dose doivent se produire à des intervalles d'au moins 1 semaine. Les patients ont été dosés dans une plage de 20 à 60 mg / jour dans les essais cliniques démontrant le efficacité de la paroxétine dans le traitement des TOC. La posologie maximale doit ne pas dépasser 60 mg / jour.
Thérapie d'entretien
Le maintien à long terme de l'efficacité a été démontré dans un Essai de prévention des rechutes de 6 mois. Dans cet essai, les patients atteints de TOC sont affectés à la paroxétine a démontré un taux de rechute inférieur à celui des patients sous placebo (voir PHARMACOLOGIE CLINIQUE). Le TOC est une maladie chronique, et c'est le cas raisonnable de considérer la poursuite pour un patient répondant. Posologie des ajustements doivent être effectués pour maintenir le patient au plus bas effectif la posologie et les patients doivent être réévalués périodiquement pour déterminer le besoin pour un traitement continu.
Trouble panique
Posologie initiale habituelle
PEXEVA® (mesylate de paroxétine) doit être administré en tant que a dose quotidienne unique avec ou sans nourriture, généralement le matin. La dose cible de paroxétine dans le traitement de la DP est de 40 mg / jour. Les patients doivent être démarrés à 10 mg / jour. Les changements de dose doivent se produire par incréments de 10 mg / jour et à intervalles d'au moins 1 semaine. Les patients ont été dosés dans une plage de 10 à 60 mg / jour dans les essais cliniques démontrant l'efficacité de la paroxétine. Le la posologie maximale ne doit pas dépasser 60 mg / jour.
Thérapie d'entretien
Le maintien à long terme de l'efficacité a été démontré dans un Essai de prévention des rechutes de 3 mois. Dans cet essai, les patients atteints de DP sont affectés à la paroxétine a démontré un taux de rechute inférieur à celui des patients sous placebo (voir PHARMACOLOGIE CLINIQUE). PD est une maladie chronique, et c'est le cas raisonnable de considérer la poursuite pour un patient répondant. Posologie des ajustements doivent être effectués pour maintenir le patient au plus bas effectif la posologie et les patients doivent être réévalués périodiquement pour déterminer le besoin pour un traitement continu.
Trouble d'anxiété généralisée
Posologie initiale habituelle
PEXEVA® (mesylate de paroxétine) doit être administré en tant que a dose quotidienne unique avec ou sans nourriture, généralement le matin. En clinique des essais sur l'efficacité de la paroxétine ont été démontrés chez des patients traités dans un plage de 20 à 50 mg / jour. La posologie de départ recommandée et la posologie établie la posologie efficace est de 20 mg / jour. Il n'y a pas de preuves suffisantes pour suggérer un plus grand bénéfice pour des doses supérieures à 20 mg / jour. Des changements de dose doivent se produire dans 10 incréments mg / jour et à des intervalles d'au moins 1 semaine.
Thérapie d'entretien
Évaluation systématique de la persistance de la paroxétine périodes pouvant aller jusqu'à 24 semaines chez les patients atteints de TAG qui avaient répondu pendant la prise la paroxétine pendant une phase de traitement aigu de 8 semaines a démontré un avantage de une telle maintenance (voir Essais cliniques). Néanmoins, les patients doivent être réévalués périodiquement pour déterminer le besoin pour le traitement d'entretien.
Populations spéciales
Traitement des femmes enceintes pendant le troisième trimestre
Néonates exposés à la paroxétine et à d'autres ISRS ou ISRS à la fin du troisième trimestre, des complications ont été prolongées hospitalisation, soutien respiratoire et alimentation par sonde (voir PRÉCAUTIONS). Lors du traitement des femmes enceintes atteintes de paroxétine au cours du troisième trimestre, le le médecin doit soigneusement examiner les risques et avantages potentiels de traitement.
Posologie pour les personnes âgées ou affaiblies, et les patients atteints de gravité Insuffisance rénale ou hépatique
La dose initiale recommandée est de 10 mg / jour pour les personnes âgées patients, patients affaiblis et / ou patients atteints d'insuffisance rénale ou hépatique sévère déficience. Des augmentations peuvent être faites si cela est indiqué. La posologie ne doit pas dépasser 40 mg / jour.
Passer un patient à ou à partir d'une monoamine oxydase Inhibiteur (IMAO) destiné à traiter les troubles psychiatriques
Au moins 14 jours devraient s'écouler entre l'arrêt de un IMAO destiné à traiter les troubles psychiatriques et l'initiation d'un traitement par PEXEVA®. Inversement, au moins 14 jours doivent être accordés après l'arrêt de PEXEVA® avant de commencer un IMAO destiné à traiter les troubles psychiatriques (voir CONTRAINDICATIONS).
Utilisation de PEXEVA® avec d'autres IMAO, tels que le linézolide ou le méthylène Bleu
Ne démarrez pas PEXEVA® chez un patient traité avec du bleu de méthylène linézolide ou intraveineux car il y a un risque accru de syndrome sérotoninergique. Chez un patient qui nécessite un traitement plus urgent d'un état psychiatrique, d'autres interventions, y compris l'hospitalisation, devraient être pris en considération (voir CONTRAINDICATIONS).
Dans certains cas, un patient recevant déjà PEXEVA® le traitement peut nécessiter un traitement urgent avec du linézolide ou du méthylène intraveineux bleu. Si des alternatives acceptables au linézolide ou au bleu de méthylène intraveineux le traitement n'est pas disponible et les avantages potentiels du linézolide ou le traitement intraveineux du bleu de méthylène est jugé supérieur aux risques de syndrome sérotoninergique chez un patient particulier, PEXEVA® doit être arrêté rapidement et du bleu de méthylène linézolide ou intraveineux peut être administré. Le patient doit être surveillé pour les symptômes du syndrome sérotoninergique pendant 2 semaines ou jusqu'à 24 ans quelques heures après la dernière dose de linézolide ou de bleu de méthylène intraveineux, selon le cas vient en premier. Le traitement par PEXEVA® peut être repris 24 heures après la dernière dose de bleu de méthylène linézolide ou intraveineux (voir AVERTISSEMENTS).
Le risque d'administrer du bleu de méthylène par voies non intraveineuses (telles que les comprimés oraux ou par injection locale) ou in les doses intraveineuses bien inférieures à 1 mg / kg avec PEXEVA® ne sont pas claires. Le le clinicien doit néanmoins être conscient de la possibilité d'émerger symptômes du syndrome sérotoninergique avec une telle utilisation (voir AVERTISSEMENTS).
Arrêt du traitement par PEXEVA® (paroxétine mésylate)
Symptômes associés à l'arrêt de la paroxétine ont été signalés (voir PRÉCAUTIONS). Les patients doivent être surveillés ces symptômes à l'arrêt du traitement, quelle que soit l'indication quelle paroxétine est prescrite. Une réduction progressive de la dose plutôt qu'un arrêt brutal est recommandé dans la mesure du possible. Si symptômes intolérables se produire après une diminution de la dose ou à l'arrêt du traitement la reprise de la dose précédemment prescrite peut être envisagée. Par la suite, le médecin peut continuer à diminuer la dose mais à un rythme plus progressif.
L'utilisation d'IMAO destinés à traiter les troubles psychiatriques avec PEXEVA® ou dans les 14 jours suivant l'arrêt du traitement par PEXEVA® contre-indiqué en raison d'un risque accru de syndrome sérotoninergique. L'utilisation de PEXEVA® dans les 14 jours suivant l'arrêt d'un IMAO destiné à traiter les psychiatriques les troubles sont également contre-indiqués (voir AVERTISSEMENTS et DOSAGE ET ADMINISTRATION).
Démarrage de PEXEVA® chez un patient traité Les IMAO tels que le linézolide ou le bleu de méthylène intraveineux sont également contre-indiqués en raison d'un risque accru de syndrome sérotoninergique (voir AVERTISSEMENTS et DOSAGE ET ADMINISTRATION).
L'utilisation concomitante chez les patients prenant de la thioridazine est contre-indiqué (voir AVERTISSEMENTS et PRÉCAUTIONS).
L'utilisation concomitante chez les patients prenant du pimozide est contre-indiqué (voir PRÉCAUTIONS).
Les comprimés de PEXEVA® (mesylate de paroxétine) sont contre-indiqués chez les patients présentant une hypersensibilité à la paroxétine ou à l'un des inactifs ingrédients des comprimés de PEXEVA® (mesylate de paroxétine).
AVERTISSEMENTS
Risque d'aggravation clinique et de suicide
Patients atteints d'un trouble dépressif majeur (TDM), tous deux adultes et pédiatrique, peut voir une aggravation de leur dépression et / ou du émergence d'idées et de comportements suicidaires (suicidalité) ou changements inhabituels comportement, qu'ils prennent ou non des antidépresseurs, et ce le risque peut persister jusqu'à ce qu'une rémission importante se produise. Le suicide est un risque connu de dépression et certains autres troubles psychiatriques, et ces troubles eux-mêmes sont les plus grands prédicteurs du suicide. Il y a une longue date craint toutefois que les antidépresseurs ne jouent un rôle dans l'aggravation de dépression et émergence de suicidalité chez certains patients au cours de la premières phases du traitement. Analyses regroupées de placebo à court terme contrôlées des essais de médicaments antidépresseurs (ISRS et autres) ont montré que ces médicaments augmenter le risque de pensée et de comportement suicidaires (suicidalité) chez les enfants adolescents et jeunes adultes (âgés de 18 à 24 ans) atteints d'un trouble dépressif majeur (TDM) et autres troubles psychiatriques. Les études à court terme n'ont pas montré d'augmentation le risque de suicidalité avec les antidépresseurs par rapport au placebo chez l'adulte au-delà de 24 ans; il y a eu une réduction avec les antidépresseurs par rapport au placebo chez les adultes de 65 ans et plus.
Les analyses regroupées des essais contrôlés contre placebo en enfants et adolescents atteints de MDD, de trouble obsessionnel compulsif (TOC), ou les autres troubles psychiatriques comprenaient un total de 24 essais à court terme sur 9 antidépresseurs chez plus de 4400 patients. Les analyses regroupées de essais contrôlés contre placebo chez des adultes atteints de MDD ou d'autres troubles psychiatriques comprenait un total de 295 essais à court terme (durée médiane de 2 mois) sur 11 antidépresseurs chez plus de 77 000 patients. Il y avait des variations considérables en risque de suicidalité chez les drogues, mais tendance à une augmentation de la patients plus jeunes pour presque tous les médicaments étudiés. Il y avait des différences dans risque absolu de suicidalité selon les différentes indications, avec le plus élevé incidence dans le MDD. Les différences de risque (médicament vs placebo), cependant, étaient relativement stable dans les strates d'âge et dans toutes les indications. Ces différences de risque (différence médicament-placebo dans le nombre de cas de suicidalité pour 1000 les patients traités) sont fournis dans le tableau 1.
TABLEAU 1
| Age Range | Différence médicament-placebo dans le nombre de cas de suicidalité pour 1000 patients traités |
| Augmente par rapport au placebo | |
| <18 | 14 cas supplémentaires |
| 18-24 | 5 cas supplémentaires |
| Diminue par rapport à Placebo | |
| 25-64 | 1 cas de moins |
| ≥ 65 | 6 cas de moins |
Aucun suicide n'est survenu dans aucun des cas essais pédiatriques. Il y a eu des suicides dans les essais pour adultes, mais le nombre l'était pas suffisant pour arriver à une conclusion sur l'effet de la drogue sur le suicide.
On ne sait pas si le le risque de suicidalité s'étend à une utilisation à plus long terme, c'est-à-dire au-delà de plusieurs mois. Toutefois il existe des preuves substantielles des essais d'entretien contrôlés contre placebo les adultes souffrant de dépression que l'utilisation d'antidépresseurs peut retarder la récurrence de dépression.
Tous les patients traités avec des antidépresseurs pour toute indication doit être surveillée de manière appropriée et observé de près pour l'aggravation clinique, la suicidalité et les changements inhabituels comportement, en particulier pendant les premiers mois d'un cours de pharmacothérapie, ou en cas de changement de dose, augmente ou diminue.
Les symptômes suivants, anxiété, agitation, panique attaques, insomnie, irritabilité, hostilité, agressivité, impulsivité, une akathisie (agitation psychomotrice), une hypomanie et une manie ont été rapportées chez les patients adultes et pédiatriques traités avec des antidépresseurs pour les majeurs trouble dépressif ainsi que pour d'autres indications, à la fois psychiatriques et non psychiatrique. Bien qu'il s'agisse d'un lien de causalité entre l'émergence de tels symptômes et soit l'aggravation de la dépression et / ou l'émergence du suicide aucune impulsion n'a été établie, on craint que de tels symptômes ne le soient représentent les précurseurs de la suicidalité émergente.
Il faudrait envisager de modifier le traitement régime, y compris l'arrêt éventuel du médicament, chez les patients dont la dépression est toujours pire ou qui connaît une suicidalité émergente ou des symptômes qui pourraient être des précurseurs d'une aggravation de la dépression ou de la suicidalité surtout si ces symptômes sont graves, brusques au début ou ne faisaient pas partie de les symptômes de présentation du patient.
Si la décision a été prise d'arrêter le traitement les médicaments doivent être effilés aussi rapidement que possible, mais avec reconnaissance cet arrêt brutal peut être associé à certains symptômes (voir PRÉCAUTIONS et DOSAGE ET ADMINISTRATION - Arrêt de Traitement par PEXEVA® (mésylate de paroxétine), pour une description des risques de arrêt de PEXEVA® (mésylate de paroxétine)).
Familles et soignants des patients traités antidépresseurs pour trouble dépressif majeur ou autres indications, les deux psychiatrique et non psychiatrique, doit être alerté sur la nécessité de surveiller patients pour l'émergence d'agitation, d'irritabilité, de changements inhabituels le comportement et les autres symptômes décrits ci-dessus, ainsi que l'émergence de suicidalité et de signaler immédiatement ces symptômes aux prestataires de soins de santé. Cette surveillance devrait inclure l'observation quotidienne des familles et des soignants. Prescriptions pour PEXEVA® (mesylate de paroxétine) doit être écrit pour la plus petite quantité de comprimés compatibles avec une bonne gestion des patients, afin de réduire le risque d'une surdose.
Dépistage des patients pour un trouble bipolaire
Un épisode dépressif majeur peut être l'initiale présentation du trouble bipolaire. On le croit généralement (mais pas établi dans des essais contrôlés) qui traitent un tel épisode avec un l'antidépresseur seul peut augmenter la probabilité de précipitation d'un épisode mixte / maniaque chez les patients à risque de trouble bipolaire. Que ce soit les symptômes décrits ci-dessus représentent une telle conversion est inconnu. Toutefois avant d'initier un traitement avec un antidépresseur, les patients dépressifs les symptômes doivent être correctement dépistés pour déterminer s'ils sont à risque trouble bipolaire; ce dépistage devrait inclure des antécédents psychiatriques détaillés y compris les antécédents familiaux de suicide, de trouble bipolaire et de dépression. Il il convient de noter que PEXEVA® (mesylate de paroxétine) n'est pas approuvé pour une utilisation dans traiter la dépression bipolaire.
Syndrome sérotoninergique
Le développement d'un danger potentiellement mortel le syndrome sérotoninergique a été signalé avec les ISRS et les ISRS, y compris PEXEVA®, seul mais surtout avec l'utilisation concomitante d'autres médicaments sérotoninergiques (y compris les triptans, les antidépresseurs tricycliques, le fentanyl, le lithium, le tramadol, tryptophane, buspirone, amphétamines et St. John's Wort) et avec des drogues qui altérer le métabolisme de la sérotonine (en particulier, les IMAO, tous deux destinés à traiter les troubles psychiatriques et autres, tels que le linézolide et intraveineux bleu de méthylène).
Les symptômes du syndrome sérotoninergique peuvent inclure un état mental changements (par ex., agitation, hallucinations, délire et coma), autonome instabilité (par ex., tachycardie, tension artérielle labile, étourdissements, diaphorèse , bouffées vasomotrices, hyperthermie), symptômes neuromusculaires (par ex., tremblements, rigidité, myoclonie, hyperréflexie, incoordination), convulsions et / ou gastro-intestinal symptômes (par ex., nausées, vomissements, diarrhée). Les patients doivent être surveillés l'émergence du syndrome sérotoninergique.
L'utilisation concomitante de PEXEVA® avec des IMAO destinés à traiter les troubles psychiatriques est contre-indiqué. PEXEVA® ne devrait pas non plus l'être a commencé chez un patient traité avec des IMAO tels que le linézolide ou bleu de méthylène intraveineux. Tous les rapports avec du bleu de méthylène fournis des informations sur la voie d'administration impliquaient une administration intraveineuse dans la plage de doses de 1 mg / kg à 8 mg / kg. Aucun rapport n'a impliqué l'administration de bleu de méthylène par d'autres voies (telles que les comprimés oraux ou l'injection tissulaire locale) ou à des doses plus faibles. Il peut y avoir des circonstances où il est nécessaire de commencer traitement avec un IMAO tel que le linezolide ou le bleu de méthylène intraveineux dans a patient prenant PEXEVA®. PEXEVA® doit être arrêté avant de commencer traitement avec l'AMI (voir CONTRAINDICATIONS et DOSAGE ET ADMINISTRATION).
En cas d'utilisation concomitante de PEXEVA® avec d'autres sérotoninergiques médicaments, y compris les triptans, les antidépresseurs tricycliques, le fentanyl, le lithium tramadol, buspirone, tryptophane, amphétamines et St. John's Wort est cliniquement justifié, soyez conscient d'un risque potentiel accru de syndrome sérotoninergique en particulier pendant l'initiation du traitement et l'augmentation de la dose.
Traitement par PEXEVA® et tout sérotoninergique concomitant les agents doivent être arrêtés immédiatement si les événements ci-dessus se produisent et un traitement symptomatique de soutien doit être instauré.
Glaucome à fermeture angulaire
La dilatation pupillaire qui se produit après l'utilisation de nombreuses personnes les antidépresseurs, y compris Pexeva, peuvent déclencher une attaque de fermeture d'angle dans a patient aux angles anatomiquement étroits qui n'a pas d'iridectomie brevetée.
Interaction potentielle avec la thioridazine
L'administration de thioridazine produit seule prolongation de l'intervalle QTc, qui est associée à une ventriculaire grave arythmies, telles que les arythmies de type torsade de pointes, et la mort subite. Cet effet semble être lié à la dose.
Une in vivo l'étude suggère que les médicaments qui inhibent Le CYP2D6, tel que la paroxétine, augmentera les taux plasmatiques de thioridazine. Par conséquent, il est recommandé de ne pas utiliser de paroxétine en association avec thioridazine (voir CONTRAINDICATIONS et PRÉCAUTIONS).
Utilisation en grossesse
Effets tératogènes
Des études épidémiologiques ont montré que les nourrissons étaient exposés à la paroxétine au premier trimestre de la grossesse ont un risque accru de malformations congénitales, en particulier les malformations cardiovasculaires. Le les résultats de ces études sont résumés ci-dessous:
- Une étude basée sur les données du registre national suédois a démontré que les nourrissons exposés à la paroxétine pendant la grossesse (n = 815) l'avaient fait un risque accru de malformations cardiovasculaires (risque de 2% en nourrissons exposés à la paroxétine) par rapport à l'ensemble de la population du registre (1% risque), pour un rapport de cotes (OR) de 1,8 (intervalle de confiance à 95% 1,1 à 2,8). Non une augmentation du risque de malformations congénitales globales a été observée dans le nourrissons exposés à la paroxétine. Les malformations cardiaques dans la paroxétine exposée les nourrissons étaient principalement des défauts septaux ventriculaires (VSD) et des septaux auriculaires défauts (TSA). Les défauts septaux varient en gravité de ceux qui résolvent spontanément à ceux qui nécessitent une intervention chirurgicale.
- Une étude de cohorte rétrospective distincte des États-Unis Les États (données de United Healthcare) ont évalué 5 956 nourrissons de mères dispensés antidépresseurs au cours du premier trimestre (n = 815 pour la paroxétine). Cette étude a montré une tendance à un risque accru de malformations cardiovasculaires paroxétine (risque de 1,5%) par rapport à d'autres antidépresseurs (risque de 1%), pour un OU de 1,5 (intervalle de confiance à 95% 0,8 à 2,9). Sur les 12 exposés à la paroxétine les nourrissons atteints de malformations cardiovasculaires, 9 avaient des VSD. Cette étude aussi a suggéré un risque accru de malformations congénitales majeures globales, y compris défauts cardiovasculaires de la paroxétine (risque de 4%) par rapport aux autres (risque de 2%) antidépresseurs (OU 1,8; intervalle de confiance à 95% 1,2 à 2,8).
- Deux grandes études cas-témoins utilisant des bases de données distinctes chacun avec> 9 000 cas d'anomalies congénitales et> 4 000 témoins, a constaté que l'utilisation maternelle de la paroxétine au cours du premier trimestre de la grossesse a été associé à un risque accru de 2 à 3 fois de sortie ventriculaire droite obstructions. Dans une étude, la RO était de 2,5 (intervalle de confiance à 95%, 1,0 à 6,0, 7 nourrissons exposés) et dans l'autre étude, la RO était de 3,3 (confiance à 95% intervalle, 1,3 à 8,8, 6 nourrissons exposés).
- D'autres études ont trouvé des résultats variables quant à savoir si il y avait un risque accru de congénitale global, cardiovasculaire ou spécifique malformations. Une méta-analyse des données épidémiologiques sur une période de 16 ans (1992 à 2008) comprenait un total de 20 études distinctes: 11 études (y compris les études susmentionnées) ont rapporté des estimations des défauts cardiovasculaires et malformations congénitales globales, 3 études ont rapporté des estimations uniquement pour les défauts cardiovasculaires et 6 études ont rapporté des estimations uniquement pour l'ensemble malformations congénitales. Bien que soumise à des limitations, cette méta-analyse a suggéré une augmentation de la fréquence des malformations cardiovasculaires (prévalence rapport de cotes [POR] 1,5; Intervalle de confiance à 95% 1,2 à 1,9) et dans l'ensemble malformations (POR 1.2; intervalle de confiance à 95% 1.1 à 1.4) avec utilisation de paroxétine au cours du premier trimestre. Ce n'était pas possible dans cette méta-analyse de déterminer dans quelle mesure les malformations cardiovasculaires pourraient avoir a contribué à des malformations globales, et il n'a pas été possible de déterminer si tout type spécifique de malformations cardiovasculaires a contribué à tous malformations cardiovasculaires.
- Si une patiente tombe enceinte en prenant de la paroxétine, elle doit être informée des dommages potentiels au fœtus. Sauf les avantages de paroxétine à la mère justifient la poursuite du traitement, il faut tenir compte être administré soit en arrêtant le traitement par paroxétine, soit en passant à un autre antidépresseur (voir PRÉCAUTIONS-Arrêt du traitement avec PEXEVA®). Pour les femmes qui ont l'intention de devenir enceintes ou qui en sont à leur première trimestre de la grossesse, la paroxétine ne doit être initiée qu'après examen des autres options de traitement disponibles.
Constatations animales
Des études de reproduction ont été réalisées à des doses allant jusqu'à 50 mg / kg / jour chez le rat et 6 mg / kg / jour chez le lapin administré pendant l'organogenèse. Ces doses sont d'environ 8 (rat) et 2 (lapin) fois le MRHD sur un mg / m² base. Ces études n'ont révélé aucune preuve d'effets tératogènes. Toutefois chez le rat, il y a eu une augmentation des décès de petits au cours des 4 premiers jours de lactation lors du dosage survenue au cours du dernier trimestre de gestation et a continué tout au long de la lactation. Cet effet s'est produit à une dose de 1 mg / kg / jour ou environ un sixième du MRHD en mg / m². La dose sans effet pour les petits de rat, la mortalité n'a pas été déterminée. La cause de ces décès ne l'est pas connu.
Effets non tératogènes
Néonates exposés au PEXEVA® et à d'autres ISRS ou sérotonine et les inhibiteurs du recaptage de la noradrénaline (IRSN), à la fin du troisième trimestre ont développé des complications nécessitant une hospitalisation prolongée, respiratoire support et alimentation par tube. De telles complications peuvent survenir immédiatement livraison. Les résultats cliniques signalés ont inclus une détresse respiratoire cyanose, apnée, convulsions, instabilité de la température, difficulté d'alimentation , vomissements, hypoglycémie, hypotonie, hypertonie, hyperréflexie, tremblements nervosité, irritabilité et pleurs constants. Ces fonctionnalités sont cohérentes avec un effet toxique direct des ISRS et des IRSN ou, éventuellement, un médicament syndrome d'arrêt. Il convient de noter que, dans certains cas, la clinique l'image est cohérente avec le syndrome sérotoninergique (voir AVERTISSEMENTS: Sérotonine Syndrome).
Les nourrissons exposés aux ISRS pendant la grossesse peuvent en avoir risque accru d'hypertension pulmonaire persistante du nouveau-né (PPHN). La PPHN survient dans 1 à 2 pour 1 000 naissances vivantes dans la population générale et l'est associée à une morbidité et une mortalité néonatales importantes. Plusieurs récents des études épidémiologiques suggèrent une association statistique positive entre les ISRS utilisation (y compris PEXEVA®) pendant la grossesse et PPHN. D'autres études ne montrent pas a association statistique importante.
Les médecins doivent également noter les résultats d'une prospective étude longitudinale de 201 femmes enceintes ayant des antécédents de dépression majeure, qui étaient soit sous antidépresseurs, soit avaient reçu des antidépresseurs inférieurs à 12 des semaines avant leur dernière période menstruelle, et étaient en rémission. Des femmes qui les médicaments antidépresseurs interrompus pendant la grossesse ont montré une importance importante augmentation de la rechute de leur dépression majeure par rapport aux femmes qui est resté sous antidépresseur tout au long de la grossesse.
Lors du traitement d'une femme enceinte avec PEXEVA®, le le médecin doit soigneusement considérer les deux risques potentiels de prendre un ISRS avec les avantages établis du traitement de la dépression avec un antidépresseur. Cette décision ne peut être prise qu'au cas par cas (voir DOSAGE ET ADMINISTRATION et RÉACTIONS INDÉSIRABLES, Rapports post-commercialisation).
PRÉCAUTIONS
Général
Activation de la manie / hypomanie
Lors des tests de pré-commercialisation, une hypomanie ou une manie s'est produite chez environ 1,0% des patients unipolaires traités par par paroxétine contre 1,1% de contrôle actif et 0,3% de patients unipolaires traités par placebo. Dans un sous-ensemble de patients classés bipolaires, le taux d'épisodes maniaques était de 2,2% paroxétine et 11,6% pour les groupes combinés de contrôle actif. Comme pour toutes les drogues efficace dans le traitement du MDD, la paroxétine doit être utilisée avec prudence patients ayant des antécédents de manie.
Convulsions
Lors des tests de pré-commercialisation, des saisies ont eu lieu dans 0,1% des cas patients traités par par paroxétine, un taux similaire à celui associé à d'autres médicaments efficace dans le traitement du MDD. La paroxétine doit être utilisée avec prudence patients ayant des antécédents de convulsions. Il doit être arrêté chez tout patient qui développe des crises.
Arrêt du traitement par PEXEVA® (paroxétine mésylate)
Essais cliniques récents soutenant les divers approuvés les indications pour la paroxétine utilisaient un régime en phase conique, plutôt qu'un arrêt brutal du traitement. Le schéma en phase conique utilisé dans GAD et Les essais cliniques sur le SSPT ont impliqué une diminution progressive de la dose quotidienne de 10 mg / jour à intervalles hebdomadaires. Lorsqu'une dose quotidienne de 20 mg / jour a été atteinte les patients ont continué cette dose pendant 1 semaine avant l'arrêt du traitement.
Avec ce régime dans ces études, les effets indésirables suivants des événements ont été signalés à une incidence de 2% ou plus pour la paroxétine à un incidence au moins deux fois celle rapportée pour le placebo: rêves anormaux , paresthésie et vertiges. Chez la majorité des patients, ces événements étaient légers à modérer et étaient spontanément limitatifs et ne nécessitaient pas d'intervention médicale.
Lors de la commercialisation de la paroxétine et d'autres ISRS et IRSN (inhibiteurs du recaptage de la sérotonine et de la noradrénaline), il y a eu spontané les rapports d'événements indésirables survenus à l'arrêt de ces médicaments (en particulier en cas de rupture), y compris les éléments suivants: humeur dysphorique, irritabilité, agitation, étourdissements, troubles sensoriels (par exemple, paresthésies telles comme sensations de choc électrique et acouphènes), anxiété, confusion, maux de tête, léthargie, labilité émotionnelle, insomnie et hypomanie. Pendant que ces événements le sont généralement spontanément résolutif, il a été signalé un arrêt grave symptômes.
Les patients doivent être surveillés pour ces symptômes lorsque arrêt du traitement par paroxétine. Une réduction progressive de la dose , plutôt qu'un arrêt brutal, est recommandé dans la mesure du possible. Si intolérable les symptômes surviennent après une diminution de la dose ou à l'arrêt de celle-ci le traitement, puis la reprise de la dose précédemment prescrite peut être envisagée. Par la suite, le médecin peut continuer à diminuer la dose mais à un plus taux progressif (voir DOSAGE ET ADMINISTRATION).
Voir aussi PRÉCAUTIONS-Utilisation pédiatrique pour les indésirables événements rapportés à l'arrêt du traitement par par paroxétine en pédiatrie patients.
Tamoxifène
Certaines études ont montré que l'efficacité du tamoxifène , tel que mesuré par le risque de rechute / mortalité du cancer du sein, peut être réduit lorsque co-prescrit avec de la paroxétine en raison de l'irréversible de la paroxétine inhibition du CYP2D6 (voir INTERACTIONS DE DROGUES). Cependant, d'autres études n'ont pas démontré un tel risque. On ne sait pas si le la co-administration de paroxétine et de tamoxifène a un effet indésirable significatif sur l'efficacité du tamoxifène. Lorsque le tamoxifène est utilisé pour le traitement ou prévention du cancer du sein, les prescripteurs devraient envisager d'utiliser une alternative antidépresseur avec peu ou pas d'inhibition du CYP2D6.
Akathisia
L'utilisation de paroxétine ou d'autres ISRS a été associée avec le développement de l'akathisie, qui se caractérise par un sens intérieur de agitation et agitation psychomoteur comme une incapacité à s'asseoir ou à se tenir debout toujours généralement associé à une détresse subjective. Cela est plus susceptible de se produire dans les premières semaines de traitement.
Hyponatrémie
Une hyponatrémie peut survenir à la suite du traitement par ISRS et IRSN, y compris PEXEVA® (mésylate de paroxétine). Dans de nombreux cas, ceci l'hyponatrémie semble être le résultat du syndrome d'inappropriation sécrétion d'hormones antidiurétiques (SIADH). Boîtes avec sodium sérique inférieur à 110 mmol / L ont été signalés. Les patients âgés peuvent être plus à risque développer une hyponatrémie avec les ISRS et les IRSN. Aussi, les patients prenant des diurétiques ou qui sont autrement épuisés en volume peut présenter un risque plus élevé (voir Gériatrique Utilisation). L'arrêt du PEXEVA® (mésylate de paroxétine) doit être envisagé chez les patients atteints d'hyponatrémie symptomatique et d'intervention médicale appropriée devrait être institué.
Les signes et symptômes de l'hyponatrémie comprennent des maux de tête difficulté à se concentrer, troubles de la mémoire, confusion, faiblesse, et imparfaitement, ce qui peut entraîner des chutes. Signes et symptômes associés à plus les cas graves et / ou aigus ont inclus hallucination, syncope, convulsions, coma arrêt respiratoire et mort.
Saignement anormal
ISRS et IRSN, y compris PEXEVA® (mésylate de paroxétine), peut augmenter le risque de saignement. Utilisation concomitante d'aspirine , les anti-inflammatoires non stéroïdiens, la warfarine et d'autres anti-coagulants peuvent ajouter à ce risque. Rapports de cas et études épidémiologiques (contrôle de cas et conception de cohorte) ont démontré une association entre l'utilisation de médicaments qui interférer avec le recaptage de la sérotonine et la survenue de gastro-intestinaux saignement. Les événements hémorragiques liés aux ISRS et aux ISRS ont varié ecchymoses, hématomes, épistaxis et pétéchies à la vie hémorragies.
Les patients doivent être avertis du risque de saignement associé à l'utilisation concomitante de PEXEVA® (mésylate de paroxétine) et AINS, aspirine ou autres médicaments qui affectent la coagulation.
Fracture osseuse
Études épidémiologiques sur le risque de fracture osseuse suivantes l'exposition à certains antidépresseurs, y compris les ISRS, a signalé une association entre le traitement antidépresseur et les fractures. Il y en a plusieurs possibles cause cette observation et on ne sait pas dans quelle mesure le risque de fracture est directement attribuable au traitement ISRS. La possibilité d'un pathologique fracture, c'est-à-dire une fracture produite par un traumatisme minimal chez un patient une diminution de la densité minérale osseuse doit être envisagée chez les patients traités par paroxétine présente une douleur osseuse inexpliquée, une sensibilité ponctuelle, un gonflement , ou des ecchymoses.
Utilisation chez les patients atteints d'une maladie concomitante
Expérience clinique avec la paroxétine chez les patients atteints certaines maladies systémiques concomitantes sont limitées. La prudence est recommandée paroxétine chez les patients atteints de maladies ou d'affections pouvant affecter le métabolisme ou réponses hémodynamiques.
La paroxétine n'a été évaluée ni utilisée par aucun étendue appréciable chez les patients ayant des antécédents récents d'infarctus du myocarde ou une maladie cardiaque instable. Les patients présentant ces diagnostics ont été exclus études cliniques lors des tests pré-marché du produit. Évaluation de électrocardiogrammes de 682 patients ayant reçu de la paroxétine en double aveugle les essais contrôlés contre placebo n'ont cependant pas indiqué que la paroxétine l'est associé au développement d'anomalies ECG importantes. De même, la paroxétine ne provoque aucun changement cliniquement important de la fréquence cardiaque ou pression artérielle.
Des concentrations plasmatiques accrues de paroxétine se produisent patients présentant une insuffisance rénale sévère (clairance de la créatinine <30 ml / min) ou insuffisance hépatique sévère. Une dose initiale plus faible doit être utilisée chez ces patients (voir DOSAGE ET ADMINISTRATION).
Informations pour les patients
PEXEVA® (mesylate de paroxétine) ne doit pas être mâché ou écrasé et doit être avalé entier.
Les patients doivent être avertis du risque de sérotonine syndrome avec l'utilisation concomitante de paroxétine et de triptans, de tramadol ou d'autres agents sérotoninergiques.
Les patients doivent être informés que la prise de Pexeva peut provoquer une légère dilatation pupillaire, qui chez les individus sensibles, peut conduire à un épisode de glaucome à fermeture d'angle. Le glaucome préexistant est presque toujours glaucome à angle ouvert car le glaucome à fermeture d'angle, lorsqu'il est diagnostiqué, peut être traité définitivement avec iridectomie. Le glaucome à angle ouvert n'est pas un facteur de risque glaucome à fermeture d'angle. Les patients peuvent souhaiter être examinés pour déterminer si ils sont sensibles à la fermeture d'angle et ont une procédure prophylactique (par ex., iridectomie), s'ils sont sensibles.
Les prescripteurs ou autres professionnels de la santé devraient en informer les patients, leurs familles et leurs soignants sur les avantages et les risques associé au traitement par PEXEVA® (mésylate de paroxétine) et doit conseiller eux dans son utilisation appropriée. Un patient Guide de médicaments sur «Antidépresseur Médicaments, dépression et autres maladies mentales graves et pensées suicidaires ou Actions »est disponible pour PEXEVA® (mesylate de paroxétine). Le prescripteur ou les professionnels de la santé devraient instruire les patients, leurs familles et leurs les soignants doivent lire le guide des médicaments et les aider à comprendre son contenu. Les patients doivent avoir la possibilité de discuter du contenu du Guide de médicaments et d'obtenir des réponses à toutes les questions qu'ils peuvent avoir. Le texte complet du Guide de médicaments est réimprimé à la fin de cela document.
Les patients doivent être informés des problèmes suivants et a demandé d'alerter leur prescripteur si ceux-ci se produisent lors de la prise de PEXEVA® (paroxétine mésylate).
Risque d'aggravation clinique et de suicide
Les patients, leurs familles et leurs soignants devraient l'être encouragé à être attentif à l'émergence de l'anxiété, de l'agitation, des crises de panique , insomnie, irritabilité, hostilité, agressivité, impulsivité, akathisie (agitation psychomotrice), hypomanie, manie, autres changements inhabituels comportement, aggravation de la dépression et idées suicidaires, surtout au début pendant le traitement antidépresseur et lorsque la dose est ajustée vers le haut ou vers le bas. Il faut conseiller aux familles et aux soignants des patients de rechercher l'émergence de tels symptômes au jour le jour, car les changements peuvent être brusques. Tel les symptômes doivent être signalés au prescripteur ou au professionnel de la santé du patient surtout s'ils sont sévères, brusques au début ou ne faisaient pas partie du les symptômes de présentation du patient. De tels symptômes peuvent être associés à un risque accru de pensée et de comportement suicidaires et indiquer un besoin de très surveillance étroite et éventuellement modifications du médicament.
Médicaments qui interfèrent avec l'hémostase (AINS, aspirine, Warfarine, etc.)
Les patients doivent être avertis de l'utilisation concomitante de paroxétine avec des AINS, de l'aspirine ou d'autres médicaments qui affectent la coagulation depuis l'utilisation combinée de médicaments psychotropes qui interfèrent avec le recaptage de la sérotonine et ces agents ont été associés à un risque accru de saignement.
Interférence avec les performances cognitives et motrices
Tout médicament psychoactif peut altérer le jugement, la pensée ou motricité. Bien que dans les études contrôlées, la paroxétine n'ait pas été démontrée altérer les performances psychomoteurs, les patients doivent être avertis de l'exploitation machines dangereuses, y compris les automobiles, jusqu'à ce qu'elles soient raisonnablement certaines que la thérapie par paroxétine n'affecte pas leur capacité à se livrer à de telles activités.
Cours complet de thérapie
Alors que les patients peuvent remarquer une amélioration avec la paroxétine thérapie dans 1 à 4 semaines, il faut leur conseiller de continuer le traitement réalisé.
Médicaments concomitants
Les patients doivent être avisés d'informer leur médecin si ils prennent ou prévoient de prendre tout médicament d'ordonnance ou de gré à gré car il existe un potentiel d'interactions.
Alcool
Bien qu'il n'ait pas été démontré que la paroxétine augmente le altération des compétences mentales et motrices causées par l'alcool, les patients doivent l'être conseillé d'éviter l'alcool lors de la prise de PEXEVA® (mésylate de paroxétine).
Grossesse
Les patients doivent être avisés d'aviser leur médecin si ils tombent enceintes ou ont l'intention de devenir enceintes pendant le traitement (voir AVERTISSEMENTS - Utilisation en grossesse: Tératogène et non tératogène Effets).
Soins infirmiers
Les patients doivent être avisés d'aviser leur médecin si ils allaitent un nourrisson (voir PRÉCAUTIONS - Mères infirmières).
Tests de laboratoire
Aucun test de laboratoire spécifique n'est recommandé.
Paxil® (chlorhydrate de paroxétine)
Paroxetine, l'ingrédient actif de PEXEVA® (paroxetine mésylate), est également l'ingrédient actif de Paxil®. Ainsi, ces deux agents ne doit pas être co-administré.
Cancérogenèse, mutagenèse, altération de la fertilité
Cancérogenèse
Des études de cancérogénicité de deux ans ont été menées en rongeurs recevant de la paroxétine dans l'alimentation à 1, 5 et 25 mg / kg / jour (souris) et 1, 5 et 20 mg / kg / jour (rats). Ces doses peuvent atteindre 2,4 (souris) et 3,9 (rat) fois la dose humaine maximale recommandée (MRHD) pour le MDD et le GAD en mg / m². Parce que le MRHD pour MDD est légèrement inférieur à celui pour les TOC (50 mg vs 60 mg), les doses utilisées dans ces études de cancérogénicité n'étaient que de 2,0 (souris) et 3,2 (rat) multiplié par le MRHD pour les TOC. Il y avait un nombre significativement plus élevé d'hommes rats du groupe à forte dose avec des sarcasmes à cellules réticulantes (1/100, 0/50, 0/50, et 4/50 pour les groupes témoins, à dose faible, moyenne et élevée, respectivement) et a tendance linéaire significativement augmentée entre les groupes pour l'occurrence de tumeurs lymphorétiques chez les rats mâles. Les rats femelles n'ont pas été affectés. Bien que il y a eu une augmentation liée à la dose du nombre de tumeurs chez la souris, il n'y en a pas eu augmentation du nombre de souris atteintes de tumeurs liée au médicament. La pertinence de ceux-ci les découvertes aux humains sont inconnues.
Mutagenèse
La paroxétine n'a produit aucun effet génotoxique dans une batterie de 5 in vitro et 2 in vivo tests qui comprenaient les éléments suivants: mutation bactérienne test, test de mutation du lymphome de souris, test de synthèse d'ADN imprévu, et tests d'aberrations cytogénétiques in vivo dans la moelle osseuse de souris et in vitro dans lymphocytes humains et dans un test létal dominant chez le rat.
Insuffisance de la fertilité
Certaines études cliniques ont montré que les ISRS (y compris paroxétine) peut affecter la qualité des spermatozoïdes pendant le traitement ISRS, ce qui peut affecter fertilité chez certains hommes.
Un taux de grossesse réduit a été trouvé dans la reproduction études chez le rat à une dose de paroxétine de 15 mg / kg / jour, soit 2,9 fois la MRHD pour MDD et GAD ou 2,4 fois le MRHD pour OCD en mg / m². Des lésions irréversibles se sont produites dans l'appareil reproducteur des rats mâles après dosage dans les études de toxicité pendant 2 à 52 semaines. Ces lésions consistaient en vacuolation de l'épithélium tubulaire épididymaire à 50 mg / kg / jour et atrophique changements dans les tubules séminifères des testicules avec spermatog arrêté
EFFETS CÔTÉ
Associé à l'arrêt du traitement
Vingt pour cent (1199/6145) des patients traités paroxétine dans les essais cliniques mondiaux en MDD et 11,8% (64/542), 9,4% (44/469) et 10,7% (79/735) des patients traités par paroxétine dans le monde les essais dans les TOC, PD et GAD, respectivement, ont interrompu le traitement en raison d'un événement indésirable. Les événements les plus courants (≥ 1%) associés à arrêt et considéré comme lié à la drogue (c.-à-d. Les événements associés) avec abandon à un taux environ deux fois ou plus pour la paroxétine par rapport au placebo) comprenait les éléments suivants:
| MDD | TOC | PD | GAD | |||||
| Paroxetine | Placebo | Paroxetine | Placebo | Paroxetine | Placebo | Paroxetine | Placebo | |
| CNS | ||||||||
| Somnolence | 2,3% | 0,7% | - | - | 1,9% | 0,3% | 2,0% | 0,2% |
| Insomnie | - | - | 1,7% | 0% | 1,3% | 0,3% | - | - |
| Agitation | 1,1% | 0,5% | - | - | - | - | - | - |
| Tremblement | 1,1% | 0,3% | - | |||||
| Vertiges | - | - | 1,5% | 0% | - | - | 1,0% | 0,2% |
| Gastro-intestinal | ||||||||
| Constipation | - | - | 1,1% | 0% | - | - | - | - |
| Nausées | 3,2% | 1,1% | 1,9% | 0% | 3,2% | 1,2% | 2,0% | 0,2% |
| Diarrhée | 1,0% | 0,3% | - | - | - | - | - | - |
| Bouche sèche | 1,0% | 0,3% | - | - | - | - | - | - |
| Vomissements | 1,0% | 0,3% | - | - | - | - | - | - |
| Autre | ||||||||
| Asthénie | 1,6% | 0,4% | 1,9% | 0,4% | - | - | 1,8% | 0,2% |
| Anormal | 1,6% | 0% | 2,1% | 0% | - | - | 2,5% | 0,5% |
| Éjaculation1 | ||||||||
| Transpiration | 1,0% | 0,3% | - | - | - | - | 1,1% | 0,2% |
| Impuissance1 | - | - | 1,5% | 0% | - | - | - | - |
| Lorsque les chiffres ne sont pas fournis, l'incidence du
les événements indésirables chez les patients traités par paroxétine n'étaient pas> 1% ou ne l'étaient pas
supérieure ou égale à deux fois l'incidence du placebo. 1 Incidence corrigée du sexe. |
||||||||
Événements indésirables couramment observés
Trouble dépressif majeur
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle pour le placebo, dérivée du tableau 2 ci-dessous) étaient: asthénie, transpiration, nausées, diminution de l'appétit , somnolence, étourdissements, insomnie, tremblements, nervosité, troubles éjaculatoires , et autres troubles génitaux masculins.
Trouble obsessionnel compulsif
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle du placebo, dérivée du tableau 3 ci-dessous) étaient: nausées, sécheresse de la bouche, diminution de l'appétit, constipation , étourdissements, somnolence, tremblements, transpiration, impuissance et éjaculation anormale.
Trouble panique
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle pour le placebo, dérivée du tableau 3 ci-dessous) étaient: asthénie, transpiration, diminution de l'appétit, libido diminution, tremblements, éjaculation anormale, troubles génitaux féminins, et impuissance.
Trouble d'anxiété généralisée
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle pour le placebo, dérivée du tableau 4) étaient: asthénie, infection, constipation, diminution de l'appétit, sec bouche, nausées, libido diminuée, somnolence, tremblements, transpiration et anormal éjaculation.
Incidence dans les essais cliniques contrôlés
Le prescripteur doit être conscient que les chiffres du les tableaux suivants ne peuvent pas être utilisés pour prédire l'incidence des effets secondaires dans le cours de pratique médicale habituelle où les caractéristiques du patient et autres les facteurs diffèrent de ceux qui ont prévalu dans les essais cliniques. De même, le les fréquences citées ne peuvent pas être comparées aux chiffres obtenus à partir d'autres cliniques enquêtes impliquant différents traitements, utilisations et enquêteurs. Le les chiffres cités fournissent cependant au médecin prescripteur une base quelconque pour estimer la contribution relative des facteurs de drogue et de non-drogue au taux d'incidence des effets secondaires dans les populations étudiées.
Trouble dépressif majeur
Le tableau 2 énumère les événements indésirables survenus à un incidence de 1% ou plus chez les patients traités par par paroxétine qui ont participé essais contrôlés versus placebo à court terme (6 semaines) dans lesquels les patients ont été dosés dans un plage de 20 à 50 mg / jour. Les événements indésirables signalés ont été classés à l'aide d'un terminologie standard du dictionnaire basé sur COSTART.
TABLEAU 2: Expérience indésirable émergente du traitement
Incidence dans les essais cliniques contrôlés par placebo pour le MDD1
| Système de carrosserie | Terme préféré | Paroxetine (n = 421) |
Placebo (n = 421) |
| Corps dans son ensemble | Maux de tête | 18% | 17% |
| Asthénie | 15% | 6% | |
| Cardiovasculaire | Palpitations | 3% | 1% |
| Vasodilatation | 3% | 1% | |
| Dermatologique | Transpiration | 11% | 2% |
| Éruption cutanée | 2% | 1% | |
| Gastro-intestinal | Nausées | 26% | 9% |
| Bouche sèche | 18% | 12% | |
| Constipation | 14% | 9% | |
| Diarrhée | 12% | 8% | |
| Diminution de l'appétit | 6% | 2% | |
| Flatulence | 4% | 2% | |
| Trouble oropharynx2 | 2% | 0% | |
| Dyspepsie | 2% | 1% | |
| Musculo-squelettique | Myopathie | 2% | 1% |
| Myalgie | 2% | 1% | |
| Myasthénie | 1% | 0% | |
| Système nerveux | Somnolence | 23% | 9% |
| Vertiges | 13% | 6% | |
| Insomnie | 13% | 6% | |
| Tremblement | 8% | 2% | |
| Nervosité | 5% | 3% | |
| Anxiété | 5% | 3% | |
| Paresthésie | 4% | 2% | |
| Libido diminuée | 3% | 0% | |
| Sentiment drogué | 2% | 1% | |
| Confusion | 1% | 0% | |
| Respiration | Bâillement | 4% | 0% |
| Sens spéciaux | Vision floue | 4% | 1% |
| Perversion gustative | 2% | 0% | |
| Système urogénital | Perturbation éjaculatoire3,4 | 13% | 0% |
| Autres troubles génitaux masculins3,5 | 10% | 0% | |
| Fréquence urinaire | 3% | 1% | |
| Trouble d'urination6 | 3% | 0% | |
| Troubles génitaux féminins3,7 | 2% | 0% | |
| 1 Événements signalés par au moins 1% des patients
traités par par la paroxétine sont inclus, sauf les événements suivants qui ont eu un
incidence sur le placebo ≥ paroxétine: douleurs abdominales, agitation, maux de dos,
douleur thoracique, stimulation du SNC, fièvre, augmentation de l'appétit, myoclonie, pharyngite ,
hypotension posturale, troubles respiratoires (comprend principalement les «symptômes froids» ou
«URI»), traumatisme et vomissements. 2 Comprend principalement «baisse dans la gorge» et «l'étanchéité dans la gorge." 3 Pourcentage corrigé pour le sexe. 4 Surtout «retard éjaculatoire." 5 Comprend «anorgasmie», «difficultés érectiles», «retardé éjaculation / orgasme »,« dysfonction sexuelle »et« impuissance." 6 Comprend principalement la «difficulté avec miction» et «urine hésitation." 7 comprend principalement «l'anorgasmie» et «la difficulté atteignant climax / orgasme." |
Trouble obsessionnel compulsif Et Trouble panique
Le tableau 3 énumère les effets indésirables événements survenus à une fréquence de 2% ou plus chez les patients atteints de TOC paroxétine qui a participé à des essais contrôlés contre placebo d'une durée de 12 semaines chez lesquels les patients ont été dosés dans une plage de 20 à 60 mg / jour ou chez les patients avec PD sur paroxétine qui a participé à des essais contrôlés contre placebo de 10 à Durée de 12 semaines pendant laquelle les patients ont été dosés dans une plage de 10 à 60 mg / jour.
TABLEAU 3: Traitement-Emergent
Incidence de l'expérience indésirable dans les essais cliniques contrôlés par placebo pour
Trouble obsessionnel compulsif et trouble panique1
| Système de carrosserie | Terme préféré | Trouble obsessionnel compulsif | Trouble panique | ||
| Paroxetine (n = 542) |
Placebo (n = 265) |
Paroxetine (n = 469) |
Placebo (n = 324) |
||
| Corps dans son ensemble | Asthénie | 22% | 14% | 14% | 5% |
| Douleur abdominale | - | - | 4% | 3% | |
| Douleur thoracique | 3% | 2% | - | - | |
| Douleur au dos | - | - | 3% | 2% | |
| Frissons | 2% | 1% | 2% | 1% | |
| Cardiovasculaire | Vasodilatation | 4% | 1% | - | - |
| Palpitations | 2% | 0% | - | - | |
| Dermatologique | Transpiration | 9% | 3% | 14% | 6% |
| Éruption cutanée | 3% | 2% | - | - | |
| Gastro-intestinal | Nausées | 23% | 10% | 23% | 17% |
| Bouche sèche | 18% | 9% | 18% | 11% | |
| Constipation | 16% | 6% | 8% | 5% | |
| Diarrhée | 10% | 10% | 12% | 7% | |
| Diminution de l'appétit | 9% | 3% | 7% | 3% | |
| Augmentation de l'appétit | 4% | 3% | 2% | 1% | |
| Système nerveux | Insomnie | 24% | 13% | 18% | 10% |
| Somnolence | 24% | 7% | 19% | 11% | |
| Vertiges | 12% | 6% | 14% | 10% | |
| Tremblement | 11% | 1% | 9% | 1% | |
| Nervosité | 9% | 8% | - | - | |
| Libido diminuée | 7% | 4% | 9% | 1% | |
| Agitation | - | - | 5% | 4% | |
| Anxiété | - | - | 5% | 4% | |
| Rêves anormaux | 4% | 1% | - | - | |
| Concentration altérée | 3% | 2% | - | - | |
| Dépersonnalisation | 3% | 0% | - | - | |
| Myoclonus | 3% | 0% | 3% | 2% | |
| Amnésie | 2% | 1% | - | - | |
| Système respiratoire | Rhinite | - | - | 3% | 0% |
| Sens spéciaux | Vision anormale | 4% | 2% | - | - |
| Perversion gustative | 2% | 0% | - | - | |
| Système urogénital | Éjaculation anormale2 | 23% | 1% | 21% | 1% |
| Trouble génital féminin2 | 3% | 0% | 9% | 1% | |
| Impuissance2 | 8% | 1% | 5% | 0% | |
| Fréquence urinaire | 3% | 1% | 2% | 0% | |
| Urisation altérée | 3% | 0% | - | - | |
| Infection par tract urinaire | 2% | 1% | 2% | 1% | |
| 1 Événements signalés par au moins 2% des TOC ou PD
les patients traités par par paroxétine sont inclus, à l'exception des événements suivants qui avaient une incidence sur le placebo ≥ paroxétine [TOC]:
douleurs abdominales, agitation, anxiété, maux de dos, toux accrue, dépression ,
maux de tête, hyperkinésie, infection, paresthésie, pharyngite, respiratoire
trouble, rhinite et sinusite. [PD]: rêves anormaux, vision anormale,
douleur thoracique, toux accrue, dépersonnalisation, dépression, dysménorrhée,
dyspepsie, syndrome grippal, maux de tête, infection, myalgie, nervosité,
palpitations, paresthésie, pharyngite, éruption cutanée, troubles respiratoires, sinusite ,
perversion gustative, traumatisme, altération de la miction et vasodilatation. 2 Pourcentage corrigé pour le sexe. |
Trouble d'anxiété généralisée
Le tableau 4 énumère les événements indésirables survenus à a fréquence de 2% ou plus chez les patients atteints de TAG sous paroxétine qui ont participé essais contrôlés contre placebo d'une durée de 8 semaines au cours desquels les patients ont été dosés dans un plage de 10 mg / jour à 50 mg / jour.
TABLEAU 4: Expérience indésirable émergente du traitement
Incidence dans les essais cliniques contrôlés par placebo pour l'anxiété généralisée
Trouble1
| Système de carrosserie | Terme préféré | Paroxetine (n = 735) |
Placebo (n = 529) |
| Corps dans son ensemble | Asthénie | 14% | 6% |
| Maux de tête | 17% | 14% | |
| Infection | 6% | 3% | |
| Cardiovasculaire | Vasodilatation | 3% | 1% |
| Dermatologique | Transpiration | 6% | 2% |
| Gastro-intestinal | Nausées | 20% | 5% |
| Bouche sèche | 11% | 5% | |
| Constipation | 10% | 2% | |
| Diarrhée | 9% | 7% | |
| Diminution de l'appétit | 5% | 1% | |
| Vomissements | 3% | 2% | |
| Système nerveux | Insomnie | 11% | 8% |
| Somnolence | 15% | 5% | |
| Vertiges | 6% | 5% | |
| Tremblement | 5% | 1% | |
| Nervosité | 4% | 3% | |
| Libido diminuée | 9% | 2% | |
| Système respiratoire | Trouble respiratoire | 7% | 5% |
| Sinusite | 4% | 3% | |
| Bâillement | 4% | - | |
| Sens spéciaux | Vision anormale | 2% | 1% |
| Système urogénital | Éjaculation anormale2 | 25% | 2% |
| Génital féminin2 | 4% | 1% | |
| Impuissance du trouble2 | 4% | 3% | |
| 1 Événements signalés par au moins 2% de GAD
les patients traités par paroxétine sont inclus, sauf les événements suivants
qui avait une incidence sur le placebo ≥ paroxétine: douleurs abdominales, dos
douleur, traumatisme, dyspepsie, myalgie et pharyngite. 2 Pourcentage corrigé pour le sexe. |
Dose Dépendance des événements indésirables
Une comparaison des événements indésirables taux dans une étude à dose fixe comparant la paroxétine 10, 20, 30 et 40 mg / jour avec le placebo dans le traitement du MDD a révélé une dépendance à la dose claire pour certains les événements indésirables les plus courants associés à l'utilisation de la paroxétine, comme indiqué dans le tableau suivant:
TABLEAU 5: Traitement-Emergent
Incidence de l'expérience indésirable dans un essai de comparaison de doses dans le traitement de
MDD *
| Système corporel / Terme préféré | Placebo n = 51 |
Paroxetine | |||
| 10 mg n = 102 |
20 mg n = 104 |
30 mg n = 101 |
40 mg n = 102 |
||
| Corps dans son ensemble | |||||
| Asthénie | 0,0% | 2,9% | 10,6% | 13,9% | 12,7% |
| Dermatologie | |||||
| Transpiration | 2,0% | 1,0% | 6,7% | 8,9% | 11,8% |
| Gastro-intestinal | |||||
| Constipation | 5,9% | 4,9% | 7,7% | 9,9% | 12,7% |
| Diminution de l'appétit | 2,0% | 2,0% | 5,8% | 4,0% | 4,9% |
| Diarrhée | 7,8% | 9,8% | 19,2% | 7,9% | 14,7% |
| Bouche sèche | 2,0% | 10,8% | 18,3% | 15,8% | 20,6% |
| Nausées | 13,7% | 14,7% | 26,9% | 34,7% | 36,3% |
| Système nerveux | |||||
| Anxiété | 0,0% | 2,0% | 5,8% | 5,9% | 5,9% |
| Vertiges | 3,9% | 6,9% | 6,7% | 8,9% | 12,7% |
| Nervosité | 0,0% | 5,9% | 5,8% | 4,0% | 2,9% |
| Paresthésie | 0,0% | 2,9% | 1,0% | 5,0% | 5,9% |
| Somnolence | 7,8% | 12,7% | 18,3% | 20,8% | 21,6% |
| Tremblement | 0,0% | 0,0% | 7,7% | 7,9% | 14,7% |
| Sens spéciaux | |||||
| Vision floue | 2,0% | 2,9% | 2,9% | 2,0% | 7,8% |
| Système urogénital | |||||
| Éjaculation anormale | 0,0% | 5,8% | 6,5% | 10,6% | 13,0% |
| Impuissance | 0,0% | 1,9% | 4,3% | 6,4% | 1,9% |
| Troubles génitaux masculins | 0,0% | 3.8 | 8,7% | 6,4% | 3,7% |
| * Règle pour inclure les effets indésirables événements du tableau: incidence d'au moins 5% pour l'un des groupes paroxétine et ≥ deux fois l'incidence du placebo pour au moins un groupe paroxétine. | |||||
Dans une étude à dose fixe comparant placebo et paroxétine 20, 40 et 60 mg dans le traitement des TOC, il n'y en avait pas relation claire entre les événements indésirables et la dose de paroxétine à laquelle les patients ont été affectés. Aucun nouvel événement indésirable n'a été observé dans la paroxétine Groupe de dose de 60 mg par rapport à l'un des autres groupes de traitement.
Dans une étude à dose fixe comparant placebo et paroxétine 10, 20 et 40 mg dans le traitement de la DP, il n'y en avait pas relation claire entre les événements indésirables et la dose de paroxétine à laquelle les patients ont été assignés, à l'exception de l'asthénie, de la bouche sèche, de l'anxiété, de la libido diminué, tremblements et éjaculation anormale. Dans les études à dose flexible, pas de nouveau des événements indésirables ont été observés chez des patients recevant 60 mg de paroxétine par rapport à l'un des autres groupes de traitement.
Dans une étude à dose fixe comparant placebo et 20 et 40 mg de paroxétine dans le traitement du GAD, pour la plupart des événements indésirables, il n'y avait pas de relation claire entre les événements indésirables et le dose de paroxétine à laquelle les patients ont été affectés, à l'exception des éléments suivants événements indésirables: asthénie, constipation et éjaculation anormale.
Dans les études de doses flexibles, non de nouveaux événements indésirables ont été observés chez des patients recevant 60 mg de paroxétine par rapport à l'un des autres groupes de traitement.
Adaptation à certains Événements indésirables: Sur une période de 4 à 6 semaines, il y avait des preuves d'adaptation à certains événements indésirables avec poursuite du traitement (par exemple, nausées et étourdissements), mais moins à d'autres effets (par exemple, bouche sèche, somnolence et asthénie).
Sexuel masculin et féminin Dysfonctionnement avec les ISRS: Bien que les changements dans le désir sexuel, les performances sexuelles, et la satisfaction sexuelle se produit souvent comme des manifestations d'un trouble psychiatrique ils peuvent également être une conséquence du traitement pharmacologique. En particulier, certains les preuves suggèrent que les ISRS peuvent provoquer de telles expériences sexuelles fâcheuses.
Estimations fiables du incidence et gravité des expériences fâcheuses impliquant le désir sexuel la performance et la satisfaction sont cependant difficiles à obtenir en partie parce que les patients et les médecins peuvent être réticents à en discuter. En conséquence, estimations de l'incidence des expériences et performances sexuelles fâcheuses citées dans l'étiquetage des produits sont susceptibles de sous-estimer leur incidence réelle.
En clinique contrôlée contre placebo essais impliquant plus de 3200 patients les plages de l'incidence rapportée des effets secondaires sexuels chez les hommes et les femmes atteints de MDD, de TOC, de DP, d'anxiété sociale le trouble, la TAG et le trouble de stress post-traumatique (SSPT) sont affichés dans le tableau 6.
TABLEAU 6: Incidence de
Événements sexuels indésirables dans les essais cliniques contrôlés
| Paroxetine | Placebo | |
| n (hommes) | 1446 | 1042 |
| Diminution de la libido | 6% -15% | 0% - 5% |
| Perturbation éjaculatoire | 13% -28% | 0% - 2% |
| Impuissance | 2% - 9% | 0% - 3% |
| n (femmes) | 1822 | 1340 |
| Diminution de la libido | 0% - 9% | 0% - 2% |
| Perturbation orgasmique | 2% - 9% | 0% - 1% |
Il n'y en a pas de adéquate et études bien contrôlées examinant la dysfonction sexuelle avec traitement par paroxétine.
Le traitement par paroxétine a été associé à plusieurs cas de priapisme. Dans les cas avec un résultat connu , les patients se sont rétablis sans séquelles.
Bien qu'il soit difficile de savoir le risque précis de dysfonctionnement sexuel associé à l'utilisation des ISRS les médecins devraient régulièrement s'enquérir de ces effets secondaires possibles.
Poids et signe vital Changements: Une perte de poids importante peut être un résultat indésirable du traitement par la paroxétine pour certains patients mais, le moyenne, les patients dans les essais contrôlés ont eu une perte de poids minimale (environ 1 livre) vs petits changements sous placebo et contrôle actif. Aucun changement significatif dans les signes vitaux (tension artérielle systolique et diastolique, pouls et température) ont été observés chez des patients traités par paroxétine en clinique contrôlée essais.
Modifications ECG: Dans une analyse des ECG obtenus en 682 patients traités par paroxétine et 415 patients traités par placebo essais cliniques contrôlés, aucun changement cliniquement significatif n'a été observé dans le ECG de chaque groupe.
Tests de la fonction hépatique: En placebo essais cliniques, les patients traités par paroxétine ont présenté des valeurs anormales tests de la fonction hépatique à un taux non supérieur à celui observé dans le placebo patients. En particulier, les comparaisons paroxétine vs placebo pour alcaline la phosphatase, la SGOT, la SGPT et la bilirubine n'ont révélé aucune différence dans le pourcentage de patients présentant des anomalies marquées.
Hallucinations: Dans les essais cliniques groupés de chlorhydrate de paroxétine à libération immédiate, des hallucinations ont été observées en 22 sur 9089 patients recevant un médicament et 4 sur 3187 patients recevant un placebo.
Autres événements observés lors de l'évaluation pré-commercialisation de Paroxetine
Lors de son évaluation pré-commercialisation en MDD, plusieurs doses de paroxétine ont été administrés à 6145 patients dans des études de phase 2 et 3. Le les conditions et la durée d'exposition à la paroxétine variaient considérablement et comprenaient (dans les catégories qui se chevauchent) études ouvertes et en double aveugle, non contrôlées et études contrôlées, études en milieu hospitalier et en ambulatoire, et dose fixe et études de titration. Au cours des essais cliniques de pré-commercialisation en TOC, PD et GAD , 542, 469 et 735 patients, respectivement, ont reçu plusieurs doses de paroxétine. Les événements indésirables associés à cette exposition ont été enregistrés par investigateurs cliniques utilisant la terminologie de leur choix. Par conséquent, il n'est pas possible de fournir une estimation significative de la proportion de les individus qui connaissent des événements indésirables sans avoir d'abord regroupé des types similaires de événements fâcheux dans un plus petit nombre de catégories d'événements standardisées.
Dans les totalisations qui suivent, les événements indésirables ont été signalés ont été classés à l'aide d'une terminologie standard du dictionnaire basé sur COSTART. Le les fréquences présentées représentent donc la proportion des 9089 patients exposé à plusieurs doses de paroxétine qui ont connu un événement du type cité au moins une fois lors de la réception de paroxétine. Tous les événements signalés sont inclus à l'exception de ceux déjà répertoriés dans les tableaux 2 à 4, ceux rapportés dans des termes si généraux qu'ils ne sont pas informatifs et des événements où un médicament cause était éloigné.
Il est important de souligner cela bien que les événements signalés lors du traitement par la paroxétine, ils ne l'étaient pas nécessairement causé par cela.
Les événements sont en outre classés par système corporel et répertoriés par ordre décroissant
Catégorie de grossesse D (voir AVERTISSEMENTS - Utilisation en grossesse: Effets tératogènes et non tératogènes).
Associé à l'arrêt du traitement
Vingt pour cent (1199/6145) des patients traités paroxétine dans les essais cliniques mondiaux en MDD et 11,8% (64/542), 9,4% (44/469) et 10,7% (79/735) des patients traités par paroxétine dans le monde les essais dans les TOC, PD et GAD, respectivement, ont interrompu le traitement en raison d'un événement indésirable. Les événements les plus courants (≥ 1%) associés à arrêt et considéré comme lié à la drogue (c.-à-d. Les événements associés) avec abandon à un taux environ deux fois ou plus pour la paroxétine par rapport au placebo) comprenait les éléments suivants:
| MDD | TOC | PD | GAD | |||||
| Paroxetine | Placebo | Paroxetine | Placebo | Paroxetine | Placebo | Paroxetine | Placebo | |
| CNS | ||||||||
| Somnolence | 2,3% | 0,7% | - | - | 1,9% | 0,3% | 2,0% | 0,2% |
| Insomnie | - | - | 1,7% | 0% | 1,3% | 0,3% | - | - |
| Agitation | 1,1% | 0,5% | - | - | - | - | - | - |
| Tremblement | 1,1% | 0,3% | - | |||||
| Vertiges | - | - | 1,5% | 0% | - | - | 1,0% | 0,2% |
| Gastro-intestinal | ||||||||
| Constipation | - | - | 1,1% | 0% | - | - | - | - |
| Nausées | 3,2% | 1,1% | 1,9% | 0% | 3,2% | 1,2% | 2,0% | 0,2% |
| Diarrhée | 1,0% | 0,3% | - | - | - | - | - | - |
| Bouche sèche | 1,0% | 0,3% | - | - | - | - | - | - |
| Vomissements | 1,0% | 0,3% | - | - | - | - | - | - |
| Autre | ||||||||
| Asthénie | 1,6% | 0,4% | 1,9% | 0,4% | - | - | 1,8% | 0,2% |
| Anormal | 1,6% | 0% | 2,1% | 0% | - | - | 2,5% | 0,5% |
| Éjaculation1 | ||||||||
| Transpiration | 1,0% | 0,3% | - | - | - | - | 1,1% | 0,2% |
| Impuissance1 | - | - | 1,5% | 0% | - | - | - | - |
| Lorsque les chiffres ne sont pas fournis, l'incidence du
les événements indésirables chez les patients traités par paroxétine n'étaient pas> 1% ou ne l'étaient pas
supérieure ou égale à deux fois l'incidence du placebo. 1 Incidence corrigée du sexe. |
||||||||
Événements indésirables couramment observés
Trouble dépressif majeur
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle pour le placebo, dérivée du tableau 2 ci-dessous) étaient: asthénie, transpiration, nausées, diminution de l'appétit , somnolence, étourdissements, insomnie, tremblements, nervosité, troubles éjaculatoires , et autres troubles génitaux masculins.
Trouble obsessionnel compulsif
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle du placebo, dérivée du tableau 3 ci-dessous) étaient: nausées, sécheresse de la bouche, diminution de l'appétit, constipation , étourdissements, somnolence, tremblements, transpiration, impuissance et éjaculation anormale.
Trouble panique
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle pour le placebo, dérivée du tableau 3 ci-dessous) étaient: asthénie, transpiration, diminution de l'appétit, libido diminution, tremblements, éjaculation anormale, troubles génitaux féminins, et impuissance.
Trouble d'anxiété généralisée
Le plus souvent observé événements indésirables associés à l'utilisation de la paroxétine (incidence de 5% ou plus élevé et incidence pour la paroxétine au moins deux fois celle pour le placebo, dérivée du tableau 4) étaient: asthénie, infection, constipation, diminution de l'appétit, sec bouche, nausées, libido diminuée, somnolence, tremblements, transpiration et anormal éjaculation.
Incidence dans les essais cliniques contrôlés
Le prescripteur doit être conscient que les chiffres du les tableaux suivants ne peuvent pas être utilisés pour prédire l'incidence des effets secondaires dans le cours de pratique médicale habituelle où les caractéristiques du patient et autres les facteurs diffèrent de ceux qui ont prévalu dans les essais cliniques. De même, le les fréquences citées ne peuvent pas être comparées aux chiffres obtenus à partir d'autres cliniques enquêtes impliquant différents traitements, utilisations et enquêteurs. Le les chiffres cités fournissent cependant au médecin prescripteur une base quelconque pour estimer la contribution relative des facteurs de drogue et de non-drogue au taux d'incidence des effets secondaires dans les populations étudiées.
Trouble dépressif majeur
Le tableau 2 énumère les événements indésirables survenus à un incidence de 1% ou plus chez les patients traités par par paroxétine qui ont participé essais contrôlés versus placebo à court terme (6 semaines) dans lesquels les patients ont été dosés dans un plage de 20 à 50 mg / jour. Les événements indésirables signalés ont été classés à l'aide d'un terminologie standard du dictionnaire basé sur COSTART.
TABLEAU 2: Expérience indésirable émergente du traitement
Incidence dans les essais cliniques contrôlés par placebo pour le MDD1
| Système de carrosserie | Terme préféré | Paroxetine (n = 421) |
Placebo (n = 421) |
| Corps dans son ensemble | Maux de tête | 18% | 17% |
| Asthénie | 15% | 6% | |
| Cardiovasculaire | Palpitations | 3% | 1% |
| Vasodilatation | 3% | 1% | |
| Dermatologique | Transpiration | 11% | 2% |
| Éruption cutanée | 2% | 1% | |
| Gastro-intestinal | Nausées | 26% | 9% |
| Bouche sèche | 18% | 12% | |
| Constipation | 14% | 9% | |
| Diarrhée | 12% | 8% | |
| Diminution de l'appétit | 6% | 2% | |
| Flatulence | 4% | 2% | |
| Trouble oropharynx2 | 2% | 0% | |
| Dyspepsie | 2% | 1% | |
| Musculo-squelettique | Myopathie | 2% | 1% |
| Myalgie | 2% | 1% | |
| Myasthénie | 1% | 0% | |
| Système nerveux | Somnolence | 23% | 9% |
| Vertiges | 13% | 6% | |
| Insomnie | 13% | 6% | |
| Tremblement | 8% | 2% | |
| Nervosité | 5% | 3% | |
| Anxiété | 5% | 3% | |
| Paresthésie | 4% | 2% | |
| Libido diminuée | 3% | 0% | |
| Sentiment drogué | 2% | 1% | |
| Confusion | 1% | 0% | |
| Respiration | Bâillement | 4% | 0% |
| Sens spéciaux | Vision floue | 4% | 1% |
| Perversion gustative | 2% | 0% | |
| Système urogénital | Perturbation éjaculatoire3,4 | 13% | 0% |
| Autres troubles génitaux masculins3,5 | 10% | 0% | |
| Fréquence urinaire | 3% | 1% | |
| Trouble d'urination6 | 3% | 0% | |
| Troubles génitaux féminins3,7 | 2% | 0% | |
| 1 Événements signalés par au moins 1% des patients
traités par par la paroxétine sont inclus, sauf les événements suivants qui ont eu un
incidence sur le placebo ≥ paroxétine: douleurs abdominales, agitation, maux de dos,
douleur thoracique, stimulation du SNC, fièvre, augmentation de l'appétit, myoclonie, pharyngite ,
hypotension posturale, troubles respiratoires (comprend principalement les «symptômes froids» ou
«URI»), traumatisme et vomissements. 2 Comprend principalement «baisse dans la gorge» et «l'étanchéité dans la gorge." 3 Pourcentage corrigé pour le sexe. 4 Surtout «retard éjaculatoire." 5 Comprend «anorgasmie», «difficultés érectiles», «retardé éjaculation / orgasme »,« dysfonction sexuelle »et« impuissance." 6 Comprend principalement la «difficulté avec miction» et «urine hésitation." 7 comprend principalement «l'anorgasmie» et «la difficulté atteignant climax / orgasme." |
Trouble obsessionnel compulsif Et Trouble panique
Le tableau 3 énumère les effets indésirables événements survenus à une fréquence de 2% ou plus chez les patients atteints de TOC paroxétine qui a participé à des essais contrôlés contre placebo d'une durée de 12 semaines chez lesquels les patients ont été dosés dans une plage de 20 à 60 mg / jour ou chez les patients avec PD sur paroxétine qui a participé à des essais contrôlés contre placebo de 10 à Durée de 12 semaines pendant laquelle les patients ont été dosés dans une plage de 10 à 60 mg / jour.
TABLEAU 3: Traitement-Emergent
Incidence de l'expérience indésirable dans les essais cliniques contrôlés par placebo pour
Trouble obsessionnel compulsif et trouble panique1
| Système de carrosserie | Terme préféré | Trouble obsessionnel compulsif | Trouble panique | ||
| Paroxetine (n = 542) |
Placebo (n = 265) |
Paroxetine (n = 469) |
Placebo (n = 324) |
||
| Corps dans son ensemble | Asthénie | 22% | 14% | 14% | 5% |
| Douleur abdominale | - | - | 4% | 3% | |
| Douleur thoracique | 3% | 2% | - | - | |
| Douleur au dos | - | - | 3% | 2% | |
| Frissons | 2% | 1% | 2% | 1% | |
| Cardiovasculaire | Vasodilatation | 4% | 1% | - | - |
| Palpitations | 2% | 0% | - | - | |
| Dermatologique | Transpiration | 9% | 3% | 14% | 6% |
| Éruption cutanée | 3% | 2% | - | - | |
| Gastro-intestinal | Nausées | 23% | 10% | 23% | 17% |
| Bouche sèche | 18% | 9% | 18% | 11% | |
| Constipation | 16% | 6% | 8% | 5% | |
| Diarrhée | 10% | 10% | 12% | 7% | |
| Diminution de l'appétit | 9% | 3% | 7% | 3% | |
| Augmentation de l'appétit | 4% | 3% | 2% | 1% | |
| Système nerveux | Insomnie | 24% | 13% | 18% | 10% |
| Somnolence | 24% | 7% | 19% | 11% | |
| Vertiges | 12% | 6% | 14% | 10% | |
| Tremblement | 11% | 1% | 9% | 1% | |
| Nervosité | 9% | 8% | - | - | |
| Libido diminuée | 7% | 4% | 9% | 1% | |
| Agitation | - | - | 5% | 4% | |
| Anxiété | - | - | 5% | 4% | |
| Rêves anormaux | 4% | 1% | - | - | |
| Concentration altérée | 3% | 2% | - | - | |
| Dépersonnalisation | 3% | 0% | - | - | |
| Myoclonus | 3% | 0% | 3% | 2% | |
| Amnésie | 2% | 1% | - | - | |
| Système respiratoire | Rhinite | - | - | 3% | 0% |
| Sens spéciaux | Vision anormale | 4% | 2% | - | - |
| Perversion gustative | 2% | 0% | - | - | |
| Système urogénital | Éjaculation anormale2 | 23% | 1% | 21% | 1% |
| Trouble génital féminin2 | 3% | 0% | 9% | 1% | |
| Impuissance2 | 8% | 1% | 5% | 0% | |
| Fréquence urinaire | 3% | 1% | 2% | 0% | |
| Urisation altérée | 3% | 0% | - | - | |
| Infection par tract urinaire | 2% | 1% | 2% | 1% | |
| 1 Événements signalés par au moins 2% des TOC ou PD
les patients traités par par paroxétine sont inclus, à l'exception des événements suivants qui avaient une incidence sur le placebo ≥ paroxétine [TOC]:
douleurs abdominales, agitation, anxiété, maux de dos, toux accrue, dépression ,
maux de tête, hyperkinésie, infection, paresthésie, pharyngite, respiratoire
trouble, rhinite et sinusite. [PD]: rêves anormaux, vision anormale,
douleur thoracique, toux accrue, dépersonnalisation, dépression, dysménorrhée,
dyspepsie, syndrome grippal, maux de tête, infection, myalgie, nervosité,
palpitations, paresthésie, pharyngite, éruption cutanée, troubles respiratoires, sinusite ,
perversion gustative, traumatisme, altération de la miction et vasodilatation. 2 Pourcentage corrigé pour le sexe. |
Trouble d'anxiété généralisée
Le tableau 4 énumère les événements indésirables survenus à a fréquence de 2% ou plus chez les patients atteints de TAG sous paroxétine qui ont participé essais contrôlés contre placebo d'une durée de 8 semaines au cours desquels les patients ont été dosés dans un plage de 10 mg / jour à 50 mg / jour.
TABLEAU 4: Expérience indésirable émergente du traitement
Incidence dans les essais cliniques contrôlés par placebo pour l'anxiété généralisée
Trouble1
| Système de carrosserie | Terme préféré | Paroxetine (n = 735) |
Placebo (n = 529) |
| Corps dans son ensemble | Asthénie | 14% | 6% |
| Maux de tête | 17% | 14% | |
| Infection | 6% | 3% | |
| Cardiovasculaire | Vasodilatation | 3% | 1% |
| Dermatologique | Transpiration | 6% | 2% |
| Gastro-intestinal | Nausées | 20% | 5% |
| Bouche sèche | 11% | 5% | |
| Constipation | 10% | 2% | |
| Diarrhée | 9% | 7% | |
| Diminution de l'appétit | 5% | 1% | |
| Vomissements | 3% | 2% | |
| Système nerveux | Insomnie | 11% | 8% |
| Somnolence | 15% | 5% | |
| Vertiges | 6% | 5% | |
| Tremblement | 5% | 1% | |
| Nervosité | 4% | 3% | |
| Libido diminuée | 9% | 2% | |
| Système respiratoire | Trouble respiratoire | 7% | 5% |
| Sinusite | 4% | 3% | |
| Bâillement | 4% | - | |
| Sens spéciaux | Vision anormale | 2% | 1% |
| Système urogénital | Éjaculation anormale2 | 25% | 2% |
| Génital féminin2 | 4% | 1% | |
| Impuissance du trouble2 | 4% | 3% | |
| 1 Événements signalés par au moins 2% de GAD
les patients traités par paroxétine sont inclus, sauf les événements suivants
qui avait une incidence sur le placebo ≥ paroxétine: douleurs abdominales, dos
douleur, traumatisme, dyspepsie, myalgie et pharyngite. 2 Pourcentage corrigé pour le sexe. |
Dose Dépendance des événements indésirables
Une comparaison des événements indésirables taux dans une étude à dose fixe comparant la paroxétine 10, 20, 30 et 40 mg / jour avec le placebo dans le traitement du MDD a révélé une dépendance à la dose claire pour certains les événements indésirables les plus courants associés à l'utilisation de la paroxétine, comme indiqué dans le tableau suivant:
TABLEAU 5: Traitement-Emergent
Incidence de l'expérience indésirable dans un essai de comparaison de doses dans le traitement de
MDD *
| Système corporel / Terme préféré | Placebo n = 51 |
Paroxetine | |||
| 10 mg n = 102 |
20 mg n = 104 |
30 mg n = 101 |
40 mg n = 102 |
||
| Corps dans son ensemble | |||||
| Asthénie | 0,0% | 2,9% | 10,6% | 13,9% | 12,7% |
| Dermatologie | |||||
| Transpiration | 2,0% | 1,0% | 6,7% | 8,9% | 11,8% |
| Gastro-intestinal | |||||
| Constipation | 5,9% | 4,9% | 7,7% | 9,9% | 12,7% |
| Diminution de l'appétit | 2,0% | 2,0% | 5,8% | 4,0% | 4,9% |
| Diarrhée | 7,8% | 9,8% | 19,2% | 7,9% | 14,7% |
| Bouche sèche | 2,0% | 10,8% | 18,3% | 15,8% | 20,6% |
| Nausées | 13,7% | 14,7% | 26,9% | 34,7% | 36,3% |
| Système nerveux | |||||
| Anxiété | 0,0% | 2,0% | 5,8% | 5,9% | 5,9% |
| Vertiges | 3,9% | 6,9% | 6,7% | 8,9% | 12,7% |
| Nervosité | 0,0% | 5,9% | 5,8% | 4,0% | 2,9% |
| Paresthésie | 0,0% | 2,9% | 1,0% | 5,0% | 5,9% |
| Somnolence | 7,8% | 12,7% | 18,3% | 20,8% | 21,6% |
| Tremblement | 0,0% | 0,0% | 7,7% | 7,9% | 14,7% |
| Sens spéciaux | |||||
| Vision floue | 2,0% | 2,9% | 2,9% | 2,0% | 7,8% |
| Système urogénital | |||||
| Éjaculation anormale | 0,0% | 5,8% | 6,5% | 10,6% | 13,0% |
| Impuissance | 0,0% | 1,9% | 4,3% | 6,4% | 1,9% |
| Troubles génitaux masculins | 0,0% | 3.8 | 8,7% | 6,4% | 3,7% |
| * Règle pour inclure les effets indésirables événements du tableau: incidence d'au moins 5% pour l'un des groupes paroxétine et ≥ deux fois l'incidence du placebo pour au moins un groupe paroxétine. | |||||
Dans une étude à dose fixe comparant placebo et paroxétine 20, 40 et 60 mg dans le traitement des TOC, il n'y en avait pas relation claire entre les événements indésirables et la dose de paroxétine à laquelle les patients ont été affectés. Aucun nouvel événement indésirable n'a été observé dans la paroxétine Groupe de dose de 60 mg par rapport à l'un des autres groupes de traitement.
Dans une étude à dose fixe comparant placebo et paroxétine 10, 20 et 40 mg dans le traitement de la DP, il n'y en avait pas relation claire entre les événements indésirables et la dose de paroxétine à laquelle les patients ont été assignés, à l'exception de l'asthénie, de la bouche sèche, de l'anxiété, de la libido diminué, tremblements et éjaculation anormale. Dans les études à dose flexible, pas de nouveau des événements indésirables ont été observés chez des patients recevant 60 mg de paroxétine par rapport à l'un des autres groupes de traitement.
Dans une étude à dose fixe comparant placebo et 20 et 40 mg de paroxétine dans le traitement du GAD, pour la plupart des événements indésirables, il n'y avait pas de relation claire entre les événements indésirables et le dose de paroxétine à laquelle les patients ont été affectés, à l'exception des éléments suivants événements indésirables: asthénie, constipation et éjaculation anormale.
Dans les études de doses flexibles, non de nouveaux événements indésirables ont été observés chez des patients recevant 60 mg de paroxétine par rapport à l'un des autres groupes de traitement.
Adaptation à certains Événements indésirables: Sur une période de 4 à 6 semaines, il y avait des preuves d'adaptation à certains événements indésirables avec poursuite du traitement (par exemple, nausées et étourdissements), mais moins à d'autres effets (par exemple, bouche sèche, somnolence et asthénie).
Sexuel masculin et féminin Dysfonctionnement avec les ISRS: Bien que les changements dans le désir sexuel, les performances sexuelles, et la satisfaction sexuelle se produit souvent comme des manifestations d'un trouble psychiatrique ils peuvent également être une conséquence du traitement pharmacologique. En particulier, certains les preuves suggèrent que les ISRS peuvent provoquer de telles expériences sexuelles fâcheuses.
Estimations fiables du incidence et gravité des expériences fâcheuses impliquant le désir sexuel la performance et la satisfaction sont cependant difficiles à obtenir en partie parce que les patients et les médecins peuvent être réticents à en discuter. En conséquence, estimations de l'incidence des expériences et performances sexuelles fâcheuses citées dans l'étiquetage des produits sont susceptibles de sous-estimer leur incidence réelle.
En clinique contrôlée contre placebo essais impliquant plus de 3200 patients les plages de l'incidence rapportée des effets secondaires sexuels chez les hommes et les femmes atteints de MDD, de TOC, de DP, d'anxiété sociale le trouble, la TAG et le trouble de stress post-traumatique (SSPT) sont affichés dans le tableau 6.
TABLEAU 6: Incidence de
Événements sexuels indésirables dans les essais cliniques contrôlés
| Paroxetine | Placebo | |
| n (hommes) | 1446 | 1042 |
| Diminution de la libido | 6% -15% | 0% - 5% |
| Perturbation éjaculatoire | 13% -28% | 0% - 2% |
| Impuissance | 2% - 9% | 0% - 3% |
| n (femmes) | 1822 | 1340 |
| Diminution de la libido | 0% - 9% | 0% - 2% |
| Perturbation orgasmique | 2% - 9% | 0% - 1% |
Il n'y en a pas de adéquate et études bien contrôlées examinant la dysfonction sexuelle avec traitement par paroxétine.
Le traitement par paroxétine a été associé à plusieurs cas de priapisme. Dans les cas avec un résultat connu , les patients se sont rétablis sans séquelles.
Bien qu'il soit difficile de savoir le risque précis de dysfonctionnement sexuel associé à l'utilisation des ISRS les médecins devraient régulièrement s'enquérir de ces effets secondaires possibles.
Poids et signe vital Changements: Une perte de poids importante peut être un résultat indésirable du traitement par la paroxétine pour certains patients mais, le moyenne, les patients dans les essais contrôlés ont eu une perte de poids minimale (environ 1 livre) vs petits changements sous placebo et contrôle actif. Aucun changement significatif dans les signes vitaux (tension artérielle systolique et diastolique, pouls et température) ont été observés chez des patients traités par paroxétine en clinique contrôlée essais.
Modifications ECG: Dans une analyse des ECG obtenus en 682 patients traités par paroxétine et 415 patients traités par placebo essais cliniques contrôlés, aucun changement cliniquement significatif n'a été observé dans le ECG de chaque groupe.
Tests de la fonction hépatique: En placebo essais cliniques, les patients traités par paroxétine ont présenté des valeurs anormales tests de la fonction hépatique à un taux non supérieur à celui observé dans le placebo patients. En particulier, les comparaisons paroxétine vs placebo pour alcaline la phosphatase, la SGOT, la SGPT et la bilirubine n'ont révélé aucune différence dans le pourcentage de patients présentant des anomalies marquées.
Hallucinations: Dans les essais cliniques groupés de chlorhydrate de paroxétine à libération immédiate, des hallucinations ont été observées en 22 sur 9089 patients recevant un médicament et 4 sur 3187 patients recevant un placebo.
Autres événements observés lors de l'évaluation pré-commercialisation de Paroxetine
Lors de son évaluation pré-commercialisation en MDD, plusieurs doses de paroxétine ont été administrés à 6145 patients dans des études de phase 2 et 3. Le les conditions et la durée d'exposition à la paroxétine variaient considérablement et comprenaient (dans les catégories qui se chevauchent) études ouvertes et en double aveugle, non contrôlées et études contrôlées, études en milieu hospitalier et en ambulatoire, et dose fixe et études de titration. Au cours des essais cliniques de pré-commercialisation en TOC, PD et GAD , 542, 469 et 735 patients, respectivement, ont reçu plusieurs doses de paroxétine. Les événements indésirables associés à cette exposition ont été enregistrés par investigateurs cliniques utilisant la terminologie de leur choix. Par conséquent, il n'est pas possible de fournir une estimation significative de la proportion de les individus qui connaissent des événements indésirables sans avoir d'abord regroupé des types similaires de événements fâcheux dans un plus petit nombre de catégories d'événements standardisées.
Dans les totalisations qui suivent, les événements indésirables ont été signalés ont été classés à l'aide d'une terminologie standard du dictionnaire basé sur COSTART. Le les fréquences présentées représentent donc la proportion des 9089 patients exposé à plusieurs doses de paroxétine qui ont connu un événement du type cité au moins une fois lors de la réception de paroxétine. Tous les événements signalés sont inclus à l'exception de ceux déjà répertoriés dans les tableaux 2 à 4, ceux rapportés dans des termes si généraux qu'ils ne sont pas informatifs et des événements où un médicament cause était éloigné.
Il est important de souligner cela bien que les événements signalés lors du traitement par la paroxétine, ils ne l'étaient pas nécessairement causé par cela.
Les événements sont en outre classés par système corporel et répertoriés par ordre décroissant de fréquence selon
Expérience humaine
Depuis l'introduction de la paroxétine aux États-Unis, 342 cas spontanés de surdosage délibéré ou accidentel pendant la paroxétine un traitement a été signalé dans le monde entier (vers 1999). Il s'agit notamment de surdoses avec de la paroxétine seule et en combinaison avec d'autres substances. De ce nombre, 48 les cas ont été mortels et, parmi les décès, 17 semblaient impliquer la paroxétine seul. Huit cas mortels qui ont documenté la quantité de paroxétine ingérée ont été généralement confondus par l'ingestion d'autres drogues ou d'alcool ou de la présence de conditions comorbides importantes. Sur 145 cas non mortels connus résultat, la plupart récupérés sans séquelles. La plus grande ingestion connue impliquée 2000 mg de paroxétine (33 fois la dose quotidienne maximale recommandée) dans a patient qui s'est rétabli.
Événements indésirables fréquemment rapportés associés le surdosage de paroxétine comprend la somnolence, le coma, les nausées, les tremblements, la tachycardie confusion, vomissements et étourdissements. Autres signes et symptômes notables observés avec des surdoses impliquant de la paroxétine (seule ou avec d'autres substances) inclure la mydriase , convulsions (y compris le statut épileptique), dysrythmies ventriculaires (y compris torsade de pointes), hypertension, réactions agressives, syncope, hypotension, stupeur, bradycardie, dystonie, rhabdomyolyse, symptômes de dysfonctionnement hépatique (y compris insuffisance hépatique, nécrose hépatique, jaunisse, hépatite et hépatique stéatose), syndrome sérotoninergique, réactions maniaques, myoclonie, rein aigu échec et rétention urinaire.
Gestion des surdosages
Aucun antidote spécifique pour la paroxétine n'est connu. Traitement devrait consister en les mesures générales utilisées dans la gestion de surdosage avec tout médicament efficace dans le traitement du MDD
Assurer des voies respiratoires, une oxygénation et une ventilation adéquates. Surveillez le rythme cardiaque et les signes vitaux. Soutien général et symptomatique des mesures sont également recommandées. L'induction des vomissements n'est pas recommandée.
En raison du grand volume de distribution de ce médicament la diurèse forcée, la dialyse, l'hémoperfusion et la transfusion d'échange sont peu probables être bénéfique.
Une prudence particulière concerne les patients qui prennent ou ont récemment pris de la paroxétine qui pourrait ingérer des quantités excessives de a antidépresseur tricyclique. Dans un tel cas, accumulation du parent tricyclique et / ou un métabolite actif peut augmenter la possibilité d'importance clinique séquelles et prolonger le temps nécessaire à une observation médicale étroite (voir Médicaments Métabolisé par le CYP2D6 du cytochrome sous PRÉCAUTIONS).
Dans la gestion du surdosage, envisagez la possibilité de implication multiple de médicaments. Le médecin devrait envisager de contacter un poison centre de contrôle pour des informations supplémentaires sur le traitement de tout surdosage. Les numéros de téléphone des centres de lutte contre les poisons certifiés sont répertoriés dans les médecins Référence de bureau (PDR).
L'efficacité de la paroxétine dans le traitement du MDD, du TOC , le trouble panique (PD) et le trouble d'anxiété généralisée (TAG) sont présumés l'être lié à la potentialisation de l'activité sérotoninergique dans le système nerveux central résultant de l'inhibition du recaptage neuronal de la sérotonine (5-hydroxytryptamine, 5-HT). Études à des doses cliniquement pertinentes chez l'homme ont démontré que la paroxétine bloque l'absorption de sérotonine en humain plaquettes. In vitro des études chez l'animal suggèrent également que la paroxétine est puissante et inhibiteur hautement sélectif du recaptage de la sérotonine neuronale et n'a que très effets faibles sur la noradrénaline et le réabsorption neuronal de la dopamine. In vitro radioligande des études contraignantes indiquent que la paroxétine a peu d'affinité pour la muscarinique alpha1-, alpha2-, bêta-adrénergique-, dopamine (D2) -, 5-HT1-, 5-HT2- et récepteurs de l'histamine (H1); antagonisme de la muscarinique, de l'histaminergique et les récepteurs alpha1-adrénergiques ont été associés à divers anticholinergiques effets sédatifs et cardiovasculaires pour d'autres médicaments psychotropes.
Parce que les puissances relatives de la paroxétine sont majeures les métabolites représentent au plus 1/50 du composé d'origine, ils le sont essentiellement inactif.
Le mésylate de paroxétine est complètement absorbé après la par voie orale dosage du sel de mésylate. Dans une étude dans laquelle des sujets masculins normaux (n = 25) a reçu 30 mg de paroxétine par jour pendant 24 jours, paroxétine à l'état d'équilibre les concentrations ont été atteintes d'environ 13 jours pour la plupart des sujets bien que cela puisse prendre beaucoup plus de temps chez un patient occasionnel. À stable état, les valeurs moyennes de Cmax, Tmax, Cmin et T½ étaient de 81,3 ng / ml (CV 41%), 8,1 h. (CV 56%), 43,2 ng / ml (CV 52%) et 33,2 h. (CV 52%), respectivement. Le les valeurs de Cmax et de Cmin à l'état d'équilibre étaient environ 7 et 10 fois supérieures à ce qui serait prédit à partir d'études à dose unique. Exposition au médicament à l'état d'équilibre basée sur l'ASC0-24 était environ 10 fois supérieur à ce qui aurait été prévu à partir des données à dose unique dans ces sujets. L'accumulation excessive est une conséquence du fait que l'une des enzymes qui métabolise la paroxétine est facilement saturable.
Dans les études de proportionnalité de dose à l'état d'équilibre impliquant patients âgés et non âgés, à des doses de 20 à 40 mg par jour pour les personnes âgées et 20 à 50 mg par jour pour les personnes âgées, une certaine non-linéarité a été observée les deux populations, reflétant à nouveau une voie métabolique saturable. En comparaison aux valeurs de Cmin après 20 mg par jour, les valeurs après 40 mg n'étaient que d'environ 2 à 3 fois supérieur au double.
Les effets des aliments sur la biodisponibilité de la paroxétine ont été étudiés chez des sujets ayant reçu une dose unique avec et sans nourriture. AUC n'a été que légèrement augmenté (6%) lorsque le médicament a été administré avec de la nourriture mais la Cmax était 29% plus élevé, tandis que le temps nécessaire pour atteindre la concentration plasmatique maximale a diminué de 6,4 heures après l'administration à 4,9 heures.
La paroxétine est largement métabolisée après la par voie orale administration. Les principaux métabolites sont des produits polaires et conjugués oxydation et méthylation, qui sont facilement éliminées. Conjugue avec l'acide glucuronique et le sulfate prédominent, et les principaux métabolites l'ont été isolé et identifié. Les données indiquent que les métabolites n'en ont pas plus 1/50 la puissance du composé d'origine à l'inhibition de l'absorption de sérotonine. Le le métabolisme de la paroxétine est accompli en partie par le cytochrome CYP2D6. La saturation de cette enzyme à des doses cliniques semble expliquer le non-linéarité de la cinétique de paroxétine avec augmentation de la dose et augmentation durée du traitement. Le rôle de cette enzyme dans le métabolisme de la paroxétine également suggère des interactions médicamenteuses potentielles (voir PRÉCAUTIONS).
Environ 64% d'une dose de 30 mg de solution buvable de la paroxétine a été excrétée dans l'urine avec 2% comme composé d'origine et 62% comme métabolites sur une période post-dosage de 10 jours. Environ 36% ont été excrétés dans le matières fécales (probablement via la bile), principalement sous forme de métabolites et moins de 1% sous forme de composé d'origine sur la période post-dosage de 10 jours.
Dans une méta-analyse de la paroxétine de 4 études réalisées en volontaires sains après administration multiple de 20 mg / jour à 40 mg / jour, mâles ne présentait pas de Cmax ou d'ASC significativement plus faible que les femelles.
Distribution
La paroxétine se distribue dans tout le corps, y compris le CNS, avec seulement 1% dans le plasma.
Liaison aux protéines
Environ 95% et 93% de la paroxétine y sont liés protéines plasmatiques à 100 ng / ml et 400 ng / ml, respectivement. Sous clinique les conditions, les concentrations de paroxétine seraient normalement inférieures à 400 ng / ml. La paroxétine ne modifie pas le in vitro liaison aux protéines de la phénytoïne ou warfarine.
Maladie rénale et hépatique
Des concentrations plasmatiques accrues de paroxétine se produisent sujets atteints d'insuffisance rénale et hépatique. Les concentrations plasmatiques moyennes dans les patients dont la clairance de la créatinine est inférieure à 30 ml / min étaient environ 4 fois plus que ce que l'on voit chez les volontaires normaux. Patients avec une clairance de la créatinine de 30 à 60 ml / min et les patients atteints d'insuffisance fonctionnelle hépatique avaient environ a Augmentation de 2 fois des concentrations plasmatiques (ASC, Cmax).
La posologie initiale doit donc être réduite patients présentant une insuffisance rénale ou hépatique sévère et une titration ascendante, si nécessaire, devrait être à intervalles accrus (voir DOSAGE ET ADMINISTRATION).
Patients âgés
Dans une étude à doses multiples chez les personnes âgées au quotidien des doses de paroxétine de 20, 30 et 40 mg, les concentrations de Cmin étaient d'environ 70% 80% de plus que les concentrations respectives de Cmin chez les sujets non âgés. Par conséquent, la posologie initiale chez les personnes âgées doit être réduite (voir DOSAGE ET ADMINISTRATION).
