

Composición:
Solicitud:
Usado en tratamiento:
Revisión médica por Fedorchenko Olga Valeryevna Última actualización de farmacia el 26.06.2023

¡Atención! ¡La información en la página es solo para profesionales médicos! ¡La información se recopila en Fuentes abiertas y puede contener errores significativos! ¡Tenga cuidado y vuelva a verificar toda la información de esta página!
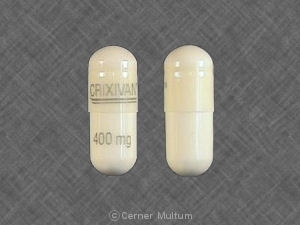
Cápsulas CRIXIVAN se suministran de la siguiente manera:
No 3756 - 200 mg cápsulas: blanco semicranslúcido. cápsulas codificadas "CRIXIVAN ™ 200 mg" en azul.
Disponible como:
NDC 0006-0571-43 botellas de unidad de uso de 360 (con desecante).
No 3758 - 400 mg cápsulas: blanco semicranslúcido. cápsulas codificadas "CRIXIVAN ™ 400 mg" en verde. Disponible como:
NDC 0006-0573-62 botellas de unidad de uso de 180 (con desecante)
Almacenamiento
Botellas: Almacenar en un recipiente herméticamente cerrado temperatura ambiente, 15-30 ° C (59-86 ° F). Proteger de la humedad.
Las cápsulas de CRIXIVAN son sensibles a la humedad. CRIXIVAN debe dispensarse y almacenarse en el contenedor original. El desecante debería permanecer en la botella original.
Dist. por: Merck Sharp & Dohme Corp., una subsidiaria de MERCK & CO., INC., Whitehouse Station, NJ 08889, EE. UU. Revisado 03/2015
CRIXIVAN en combinación con agentes antirretrovirales es indicado para el tratamiento de la infección por VIH. Esta indicación se basa en dos ensayos clínicos de aproximadamente 1 año de duración que demostraron: 1) una reducción en riesgo de enfermedades que definen el SIDA o muerte; 2) una supresión prolongada de ARN del VIH .
La dosis recomendada de CRIXIVAN es de 800 mg (generalmente dos Cápsulas de 400 mg) por vía oral cada 8 horas.
CRIXIVAN debe tomarse a intervalos de 8 horas. Por absorción óptima, CRIXIVAN debe administrarse sin alimentos pero con agua 1 hora antes o 2 horas después de una comida. Alternativamente, CRIXIVAN puede ser administrado con otros líquidos como leche descremada, jugo, café o té, o con una comida ligera, p., tostadas secas con gelatina, jugo y café con leche descremada y azúcar; o copos de maíz, leche descremada y azúcar. (Ver FARMACOLOGÍA CLÍNICA, Efecto de los alimentos sobre la absorción oral.)
Para garantizar una hidratación adecuada, se recomienda que los adultos beben al menos 1,5 litros (aproximadamente 48 onzas) de líquidos durante el período El curso de 24 horas.
Terapia concomitante (Ver FARMACOLOGÍA CLÍNICA, Interacciones farmacológicas, y / o PRECAUCIONES : INTERACCIONES DE DROGAS.)
Delavirdina
La reducción de la dosis de CRIXIVAN a 600 mg cada 8 horas debería ser considerado al administrar delavirdina 400 mg tres veces al día.
Didanosina
Si se administran indinavir y didanosina concomitantemente, deben administrarse con al menos una hora de diferencia en un vacío estómago (consulte la circular del producto del fabricante para didanosina).
Itraconazol
La reducción de la dosis de CRIXIVAN a 600 mg cada 8 horas es recomendado al administrar itraconazol 200 mg dos veces al día simultáneamente.
Ketoconazol
La reducción de la dosis de CRIXIVAN a 600 mg cada 8 horas es recomendado al administrar ketoconazol simultáneamente.
Rifabutina
Reducción de dosis de rifabutina a la mitad de la dosis estándar (consulte la circular del producto del fabricante para rifabutina) y un aumento de la dosis de CRIXIVAN a 1000 mg cada 8 horas se recomiendan cuando rifabutina y CRIXIVAN se coadministran.
Insuficiencia hepática
La dosis de CRIXIVAN debe reducirse a 600 mg cada una 8 horas en pacientes con insuficiencia hepática leve-tomoderata debido a cirrosis.
Nefrolitiasis / Urolitiasis
Además de la hidratación adecuada, la gestión médica en Los pacientes que experimentan nefrolitiasis / urolitiasis pueden incluir temporal interrupción (p. ej., 1 a 3 días) o interrupción de la terapia.
CRIXIVAN está contraindicado en pacientes con clínica hipersensibilidad significativa a cualquiera de sus componentes.
La inhibición de CYP3A4 por CRIXIVAN puede resultar en elevada concentraciones plasmáticas de los siguientes fármacos, que potencialmente causan graves o reacciones potencialmente mortales :
Tabla 7: Interacciones farmacológicas con Crixivan :
Drogas contraindicadas
| Clase de drogas | Drogas dentro de la clase que están contraindicadas con CRIXIVAN |
| Antagonista del receptor alfa 1-adrenoreceptor | alfuzosina |
| Antiarrítmicos | amiodarona |
| Derivados de ergot | dihidroergotamina, ergonovina, ergotamina, metilergonovina |
| Agentes de motilidad GI | cisaprida |
| Inhibidores de la HMG-CoA Reductasa | lovastatina, simvastatina |
| Neurolépticos | pimozida |
| Inhibidores de PDE5 | Revatio (sildenafil) [para el tratamiento de la hipertensión arterial pulmonar] |
| Sedante / hipnótico | midazolam oral, triazolam, alprazolam |
| * Marca registrada de Pfizer, Inc. |
ADVERTENCIAS
ALERTA: Conozca los medicamentos que NO deberían ser tomado con CRIXIVAN . Esta declaración está incluida en la botella del producto etiqueta.
Nefrolitiasis / Urolitiasis
La nefrolitiasis / urolitiasis ha ocurrido con CRIXIVAN terapia. La frecuencia acumulada de nefrolitiasis es sustancialmente mayor en pacientes pediátricos (29%) que en pacientes adultos (12.4%; rango en individuos ensayos: 4.7% a 34.4%). La frecuencia acumulativa de eventos de nefrolitiasis aumenta con el aumento de la exposición a CRIXIVAN; sin embargo, el riesgo a lo largo del tiempo permanece relativamente constante. En algunos casos, la nefrolitiasis / urolitiasis tiene se ha asociado con insuficiencia renal o insuficiencia renal aguda, pielonefritis con o sin bacteriemia. Si signos o síntomas de Se produce nefrolitiasis / urolitiasis (incluido el dolor en el flanco, con o sin hematuria o hematuria microscópica), interrupción temporal (p. ej., 1-3 días) o se puede considerar la interrupción de la terapia. La hidratación adecuada es recomendado en todos los pacientes tratados con CRIXIVAN. (Ver REACCIONES ADVERSAS y DOSIS Y ADMINISTRACIÓN, Nefrolitiasis / Urolitiasis.)
Anemia hemolítica
Anemia hemolítica aguda, incluidos los casos que resultan muerte, se ha informado en pacientes tratados con CRIXIVAN. Una vez que un diagnóstico es deben ser medidas aparentes y apropiadas para el tratamiento de la anemia hemolítica instituido, incluida la interrupción de CRIXIVAN
Hepatitis
Hepatitis, incluidos los casos que resultan en insuficiencia hepática y se ha informado la muerte en pacientes tratados con CRIXIVAN. Porque el La mayoría de estos pacientes tenían condiciones médicas confusas y / o estaban recibiendo terapia (s) concomitante (s), una relación causal entre CRIXIVAN y estos Los eventos no se han establecido.
Hiperglucemia
Diabetes mellitus de nueva aparición, exacerbación de preexistente Se han informado diabetes mellitus e hiperglucemia durante la postcomercialización vigilancia en pacientes infectados por VIH que reciben terapia con inhibidores de la proteasa. Algunos pacientes requirieron iniciación o ajustes de dosis de insulina u oral agentes hipoglucemiantes para el tratamiento de estos eventos. En algunos casos, diabético Se ha producido cetoacidosis. En aquellos pacientes que descontinuaron la proteasa terapia con inhibidores, la hiperglucemia persistió en algunos casos. Porque estos eventos se han informado voluntariamente durante la práctica clínica, estimaciones de frecuencia no se puede hacer y una relación causal entre la terapia con inhibidores de la proteasa y Estos eventos no se han establecido.
Riesgo de reacciones adversas graves debido al medicamento Interacciones
Iniciación de CRIXIVAN, un inhibidor de CYP3A, en pacientes recibir medicamentos metabolizados por CYP3A o inicio de medicamentos metabolizado por CYP3A en pacientes que ya reciben CRIXIVAN, puede aumentar el plasma concentraciones de medicamentos metabolizados por CYP3A. Iniciación de medicamentos que inhibe o induce CYP3A puede aumentar o disminuir las concentraciones de CRIXIVAN, respectivamente. Estas interacciones pueden conducir a:
- Reacciones adversas clínicamente significativas, potencialmente que conduce a eventos severos, potencialmente mortales o fatales de mayores exposiciones de medicamentos concomitantes.
- Reacciones adversas clínicamente significativas de mayores exposiciones de CRIXIVAN .
- Pérdida del efecto terapéutico de CRIXIVAN y posible desarrollo de resistencia.
Consulte la Tabla 9 para conocer los pasos para prevenir o administrar estos posibles y interacciones farmacológicas significativas conocidas, incluidas las recomendaciones de dosificación. Considere el potencial de interacciones farmacológicas antes y durante CRIXIVAN terapia; revisar medicamentos concomitantes durante la terapia con CRIXIVAN; y monitor para las reacciones adversas asociadas con los medicamentos concomitantes.
Uso concomitante de CRIXIVAN con lovastatina o la simvastatina está contraindicada debido a un mayor riesgo de miopatía, incluido rabdomiólisis. Se debe tener precaución si CRIXIVAN se usa simultáneamente con atorvastatina o rosuvastatina. Titule la atorvastatina y la rosuvastatina dosifica cuidadosamente y usa la dosis más baja necesaria con CRIXIVAN. (Ver PRECAUCIONES : INTERACCIONES DE DROGAS.)
El midazolam se metaboliza ampliamente por CYP3A4. La administración conjunta con CRIXIVAN con o sin ritonavir puede causar una gran cantidad aumento en la concentración de esta benzodiacepina. No hay estudio de interacción farmacológica se ha realizado para la administración conjunta de CRIXIVAN con benzodiacepinas. Según los datos de otros inhibidores de CYP3A4, las concentraciones plasmáticas de midazolam se espera que sea significativamente mayor cuando se administra midazolam por vía oral. Por lo tanto, CRIXIVAN no debe administrarse conjuntamente con administración oral midazolam (ver CONTRAINDICACIONES), mientras que se debe tener precaución administración conjunta de CRIXIVAN y midazolam parenteral. Datos de concomitante El uso de midazolam parenteral con otros inhibidores de la proteasa sugiere una posible Aumento de 3-4 veces en los niveles plasmáticos de midazolam. Si CRIXIVAN con o sin ritonavir se administra conjuntamente con midazolam parenteral, debe hacerse en a entorno que garantiza una estrecha vigilancia clínica y un examen médico adecuado manejo en caso de depresión respiratoria y / o sedación prolongada. Dosis Se debe considerar la reducción de midazolam, especialmente si es más de un solo se administra la dosis de midazolam.
Se debe tener especial precaución al prescribir sildenafil, tadalafil o vardenafil en pacientes que reciben indinavir. Se espera que la administración conjunta de CRIXIVAN con estos medicamentos Aumentar sustancialmente las concentraciones plasmáticas de sildenafil, tadalafil y vardenafil y puede provocar un aumento de los eventos adversos, incluidos hipotensión, cambios visuales y priapismo, que se han asociado con sildenafil tadalafil y vardenafil (ver CONTRAINDICACIONES y PRECAUCIONES : INTERACCIONES DE DROGAS y INFORMACIÓN PACIENTE, y el información de prescripción completa del fabricante para sildenafil, tadalafil o vardenafil).
Uso concomitante de CRIXIVAN y St. Hierba de John (Hypericum perforatum) o productos que contienen St. La hierba de John no se recomienda. Administración conjunta de CRIXIVAN y St. Se ha demostrado que la hierba de John disminuye sustancialmente concentraciones de indinavir (ver FARMACOLOGÍA CLÍNICA, DROGAS INTERACCIONES) y puede conducir a la pérdida de respuesta virológica y posible resistencia a CRIXIVAN o a la clase de inhibidores de la proteasa.
PRECAUCIONES
General
La hiperbilirrubinemia indirecta ha ocurrido con frecuencia durante el tratamiento con CRIXIVAN y con poca frecuencia se ha asociado con él aumentos en las transaminasas séricas (ver también REACCIONES ADVERSAS, Clínico Ensayos y Experiencia posterior a la comercialización). No se sabe si CRIXIVAN exacerbará la hiperbilirrubinemia fisiológica observada en los recién nacidos. (Ver Embarazo.)
Nefritis tubulointersticial
Informes de nefritis tubulointersticial con medular Se han observado calcificación y atrofia cortical en pacientes con leucocituria grave asintomática (> 100 células / campo de alta potencia). Pacientes con leucocituria severa asintomática debe seguirse de cerca y controlarse frecuentemente con urinarios. Se puede justificar una evaluación diagnóstica adicional, y La interrupción de CRIXIVAN debe considerarse en todos los pacientes con enfermedad grave leucocituria.
Se ha informado el síndrome de reconstitución inmune en pacientes tratados con terapia antirretroviral combinada, incluido CRIXIVAN Durante la fase inicial del tratamiento antirretroviral combinado, pacientes de los cuales El sistema inmunitario responde puede desarrollar una respuesta inflamatoria a indolente o infecciones oportunistas residuales (como Mycobacterium avium infección, citomegalovirus, Pneumocystis jirovecii neumonía [PCP], o tuberculosis), que puede requerir una evaluación y tratamiento adicionales. Autoinmune trastornos (como la enfermedad de Graves, la polimiositis y el síndrome de Guillain-Barré) también se ha informado que ocurre en el contexto de la reconstitución inmune; sin embargo, El tiempo de inicio es más variable y puede ocurrir muchos meses después del inicio de tratamiento.
Condiciones coexistentes
Pacientes con hemofilia: Ha habido informes de hemorragia espontánea en pacientes con hemofilia A y B tratados con inhibidores de la proteasa. En algunos pacientes, se requirió factor VIII adicional. En muchos de los casos reportados, el tratamiento con inhibidores de la proteasa continuó o reiniciado. Una relación causal entre la terapia con inhibidores de la proteasa y estos Los episodios no se han establecido. (Ver REACCIONES ADVERSAS, Postcomercialización Experiencia.)
Pacientes con insuficiencia hepática debido a cirrosis: En estos pacientes, la dosis de CRIXIVAN debe reducirse debido a disminución del metabolismo de CRIXIVAN (ver DOSIS Y ADMINISTRACIÓN). Pacientes con insuficiencia renal: los pacientes con insuficiencia renal no lo han sido estudiado.
Redistribución gorda
Redistribución / acumulación de grasa corporal, incluida la central obesidad, agrandamiento de la grasa dorsocervical (joroba de búfalo), desgaste periférico han sido desperdicios faciales, aumento de senos y "apariencia cushingoide" observado en pacientes que reciben terapia antirretroviral. El mecanismo y Las consecuencias a largo plazo de estos eventos son actualmente desconocidas. Una causalidad La relación no se ha establecido.
Información para pacientes
Una declaración para pacientes y proveedores de atención médica es incluido en la etiqueta de la botella del producto. ALERTA: Conozca los medicamentos que NO debe tomarse con CRIXIVAN Un inserto de paquete de paciente (PPI) para CRIXIVAN está disponible para información del paciente.
CRIXIVAN no es una cura para la infección por VIH-1 y los pacientes puede continuar experimentando enfermedades asociadas con la infección por VIH-1, incluyendo infecciones oportunistas. Los pacientes deben permanecer bajo el cuidado de un médico cuando se usa CRIXIVAN .
Se debe aconsejar a los pacientes que eviten hacer cosas que puedan propagar la infección por VIH-1 a otros.
- No comparta agujas u otros equipos de inyección.
- No comparta artículos personales que puedan tener sangre o fluidos corporales sobre ellos, como cepillos de dientes y cuchillas de afeitar.
- No tenga ningún tipo de sexo sin protección. Siempre practique sexo seguro usando un condón de látex o poliuretano para reducir la posibilidad de contacto sexual con semen, secreciones vaginales o sangre.
- No amamante. No sabemos si CRIXIVAN puede pasar a su bebé en la leche materna y si podría dañar a su bebé. Además, las madres con VIH-1 no deben amamantar porque se puede transmitir el VIH-1 El bebé en la leche materna.
Se debe aconsejar a los pacientes que permanezcan bajo el cuidado de a médico cuando usa CRIXIVAN y no debe modificar o suspender el tratamiento sin consultar primero al médico. Por lo tanto, si se omite una dosis, pacientes debe tomar la siguiente dosis a la hora programada regularmente y no debe duplicarse esta dosis. La terapia con CRIXIVAN debe iniciarse y mantenerse en el dosis recomendada.
CRIXIVAN puede interactuar con algunas drogas; por lo tanto, Se debe aconsejar a los pacientes que informen a su médico sobre el uso de cualquier otro medicamentos recetados, sin receta o productos herbales, particularmente St. John's mosto.
Para una absorción óptima, se debe administrar CRIXIVAN sin comida pero con agua 1 hora antes o 2 horas después de una comida. Alternativamente, CRIXIVAN puede administrarse con otros líquidos como la leche descremada jugo, café o té, o con una comida ligera, p., tostadas secas con gelatina, jugo, y café con leche descremada y azúcar; o copos de maíz, leche descremada y azúcar (ver FARMACOLOGÍA CLÍNICA, Efecto de los alimentos sobre la absorción oral y DOSIS Y ADMINISTRACIÓN). Ingestión de CRIXIVAN con una comida rica en calorías, grasas y la proteína reduce la absorción de indinavir.
Pacientes que reciben una fosfodiesterasa tipo 5 (PDE5) se debe informar a los inhibidores (sildenafil, tadalafil o vardenafil) que ellos puede tener un mayor riesgo de eventos adversos asociados con el inhibidor de PDE5, incluidos hipotensión, cambios visuales y priapismo, y debe informar de inmediato sobre cualquier síntomas a sus médicos (ver CONTRAINDICACIONES y ADVERTENCIAS, DROGAS INTERACCIONES).
Se debe informar a los pacientes que la redistribución o La acumulación de grasa corporal puede ocurrir en pacientes que reciben terapia antirretroviral y que se desconocen la causa y los efectos a largo plazo en la salud de estas afecciones en este momento.
Las cápsulas de CRIXIVAN son sensibles a la humedad. Pacientes debe informarse que CRIXIVAN debe almacenarse y utilizarse en el original el contenedor y el desecante deben permanecer en la botella.
Carcinogénesis, mutagénesis, deterioro de la fertilidad
Se realizaron estudios de carcinogenicidad en ratones y ratas. En ratones, no se observó una mayor incidencia de ningún tipo de tumor. El más alto la dosis probada en ratas fue de 640 mg / kg / día; a esta dosis un estadísticamente significativo La mayor incidencia de adenomas tiroideos se observó solo en ratas macho. En eso dosis, la exposición sistémica diaria en ratas fue aproximadamente 1.3 veces mayor que exposición sistémica diaria en humanos. No hay evidencia de mutagenicidad o genotoxicidad fue observado en in vitro pruebas de mutagénesis microbiana (Ames) in vitro alcalino ensayos de elución para la rotura del ADN in vitro y in vivo aberración cromosómica estudios y in vitro ensayos de mutagénesis de células de mamíferos. No relacionado con el tratamiento Se observaron efectos sobre el apareamiento, la fertilidad o la supervivencia de embriones en ratas hembras y no se observaron efectos relacionados con el tratamiento sobre el rendimiento del apareamiento en ratas macho en dosis que proporcionan exposición sistémica comparable o ligeramente superior a la de con la dosis clínica. Además, no se observaron efectos relacionados con el tratamiento en fecundidad o fertilidad de hembras no tratadas apareadas con machos tratados.
Embarazo
Embarazo Categoría C: Toxicidad del desarrollo Se realizaron estudios en conejos (a dosis de hasta 240 mg / kg / día), perros (a dosis de hasta 80 mg / kg / día) y ratas (a dosis de hasta 640 mg / kg / día). El más alto Las dosis en estos estudios produjeron exposiciones sistémicas en estas especies comparables o ligeramente mayor que la exposición humana. No hay tratamiento externo relacionado Se observaron cambios viscerales o esqueléticos en conejos o perros. No Se observaron cambios externos o viscerales relacionados con el tratamiento en ratas. Aumentos relacionados con el tratamiento sobre los controles en la incidencia de costillas supernumerarias (a exposiciones iguales o inferiores a las de los humanos) y de costillas cervicales (a exposiciones comparable o ligeramente mayor que los de los humanos) se observaron en ratas. En las tres especies, sin efectos relacionados con el tratamiento sobre la supervivencia embrionaria / fetal o se observaron pesos fetales.
En conejos, a una dosis materna de 240 mg / kg / día, sin drogas se detectó en plasma fetal 1 hora después de la dosificación. Niveles de fármaco en plasma fetal 2 horas después de la dosificación fueron aproximadamente el 3% de los niveles de drogas en plasma materno. En perros, a una dosis materna de 80 mg / kg / día, los niveles de fármaco en plasma fetal fueron aproximadamente el 50% de los niveles de drogas en plasma materno, tanto 1 como 2 horas después dosificación. En ratas, a dosis maternas de 40 y 640 mg / kg / día, fármaco plasmático fetal los niveles fueron aproximadamente del 10 al 15% y del 10 al 20% de la droga plasmática materna niveles 1 y 2 horas después de la dosificación, respectivamente.
Indinavir se administró a los monos Rhesus durante el tercer trimestre del embarazo (a dosis de hasta 160 mg / kg dos veces al día) y hasta monos Rhesus neonatales (a dosis de hasta 160 mg / kg dos veces al día). Cuando se administra a los recién nacidos, indinavir causó una exacerbación de la hiperbilirrubinemia fisiológica transitoria visto en esta especie después del nacimiento; Los valores séricos de bilirrubina fueron aproximadamente cuatro veces por encima de los controles a 160 mg / kg dos veces al día. Una exacerbación similar lo hizo no ocurre en neonatos después de la exposición en el útero a indinavir durante el tercero trimestre del embarazo. En los monos Rhesus, los niveles de drogas en plasma fetal fueron aproximadamente del 1 al 2% de los niveles de drogas en plasma materno aproximadamente 1 hora después dosificación materna a 40, 80 o 160 mg / kg dos veces al día.
Se ha producido hiperbilirrubinemia durante el tratamiento con CRIXIVAN (ver PRECAUCIONES y REACCIONES ADVERSAS). Se desconoce si CRIXIVAN administrado a la madre en el período perinatal exacerbará hiperbilirrubinemia fisiológica en neonatos.
No hay estudios adecuados y bien controlados en pacientes embarazadas. CRIXIVAN debe usarse durante el embarazo solo si el El beneficio potencial justifica el riesgo potencial para el feto.
Una dosis de CRIXIVAN de 800 mg cada 8 horas (con zidovudina Se han estudiado 200 mg cada 8 horas y lamivudina 150 mg dos veces al día en 16 infectados por el VIH pacientes embarazadas a las 14 a 28 semanas de gestación al momento de la inscripción (estudio PACTG 358). Dadas las exposiciones anteparto sustancialmente más bajas observadas y las limitadas datos en esta población de pacientes, no se recomienda el uso de indinavir en Pacientes embarazadas infectados por el VIH (ver FARMACOLOGÍA CLÍNICA, Embarazada Pacientes).
Registro de embarazo antirretroviral
Monitorear los resultados materno-fetales de las pacientes embarazadas expuesto a CRIXIVAN, se ha establecido un Registro de embarazo antirretroviral. Se alienta a los médicos a registrar pacientes llamando al 1-800-258-4263.
Madres lactantes
Los estudios en ratas lactantes lo han demostrado indinavir se excreta en la leche. Aunque no se sabe si CRIXIVAN es excretado en la leche humana, existe el potencial de efectos adversos indinavir en lactantes. Se debe indicar a las madres que suspendan enfermería si están recibiendo CRIXIVAN. Esto es consistente con el recomendación de los Centros de Control de Enfermedades del Servicio de Salud Pública de EE. UU y Prevención de que las madres infectadas por el VIH no amamanten a sus bebés para evitar arriesgándose a la transmisión postnatal del VIH
Uso pediátrico
El régimen de dosificación óptimo para el uso de indinavir en pacientes pediátricos no se ha establecido. Una dosis de 500 mg / m² cada ocho Se han estudiado horas en estudios no controlados de 70 niños, de 3 a 18 años edad. Los perfiles farmacocinéticos de indinavir a esta dosis no fueron comparables a perfiles previamente observados en adultos que reciben la dosis recomendada (ver FARMACOLOGÍA CLÍNICA, Pediátrico). Aunque la supresión viral fue observado en algunos de los 32 niños que fueron seguidos en este régimen 24 semanas, se informó una tasa sustancialmente mayor de nefrolitiasis cuando en comparación con los datos históricos de adultos (ver ADVERTENCIAS, Nefrolitiasis / Urolitiasis). Médicos que consideran el uso de indinavir en pacientes pediátricos sin otro Las opciones de inhibidores de la proteasa deben conocer los datos limitados disponibles en esta población y el mayor riesgo de nefrolitiasis.
Uso geriátrico
Los estudios clínicos de CRIXIVAN no incluyeron suficientes cantidad de sujetos de 65 años o más para determinar si responden diferente de los sujetos más jóvenes. En general, selección de dosis para un anciano el paciente debe ser cauteloso, lo que refleja la mayor frecuencia de disminución función hepática, renal o cardíaca y de enfermedad concomitante u otro fármaco terapia.
(también ver CONTRAINDICACIONES, ADVERTENCIAS, PRECAUCIONES : INTERACCIONES DE DROGAS) Indinavir es un inhibidor de la isoforma CYP3A4 del citocromo P450. Administración conjunta de CRIXIVAN y los fármacos metabolizados principalmente por CYP3A4 pueden aumentar el plasma concentraciones de la otra droga, que podrían aumentar o prolongar su efectos terapéuticos y adversos (ver CONTRAINDICACIONES y ADVERTENCIAS). Basado en in vitro datos en microsomas hepáticos humanos, indinavir no inhibe CYP1A2, CYP2C9, CYP2E1 y CYP2B6. Sin embargo, indinavir puede ser un inhibidor débil de CYP2D6.
Indinavir se metaboliza por CYP3A4. Drogas que inducen Se espera que la actividad de CYP3A4 aumente el aclaramiento de indinavir resultando en concentraciones plasmáticas reducidas de indinavir. Administración conjunta de CRIXIVAN y otros medicamentos que inhiben CYP3A4 pueden disminuir el aclaramiento de indinavir y puede provocar un aumento de las concentraciones plasmáticas de indinavir.
Se realizaron estudios de interacción farmacológica con CRIXIVAN y otros medicamentos que probablemente se administren conjuntamente y algunos medicamentos comúnmente utilizados como sondas para interacciones farmacocinéticas. Los efectos de la administración conjunta de CRIXIVAN en el AUC, Cmax y Cmin se resumen en la Tabla 2 (efecto de otros medicamentos en indinavir) y Tabla 3 (efecto de indinavir en otros medicamentos). Para información con respecto a las recomendaciones clínicas, ver Tabla 9 en PRECAUCIONES.
Tabla 2: Interacciones farmacológicas: parámetros farmacocinéticos
para Indinavir en presencia del fármaco coadministrado (ver PRECAUCIONES, tabla
9 para alteraciones recomendadas en dosis o régimen)
| Droga coadministrada | Dosis de fármaco coadministrado (mg) | Dosis de CRIXIVAN (mg) | n | Relación (con / sin fármaco coadministrado) de parámetros farmacocinéticos de indinavir (90% CI); Sin efecto = 1.00 |
||
| Cmax | AUC | Cmin | ||||
| Cimetidina | 600 dos veces al día, 6 días | 400 dosis única | 12 | 1.07 (0.77, 1.49) |
0,98 (0.81, 1.19) |
0,82 (0,69, 0,99) |
| Claritromicina | 500 q12h, 7 días | 800 tres veces al día, 7 días | 10 | 1.08 (0.85, 1.38) |
1.19 (1.00, 1.42) |
1.57 (1.16, 2.12) |
| Delavirdina | 400 tres veces al día | 400 tres veces al día, 7 días | 28 | 0,64 * (0.48, 0.86) |
Sin cambios significativos * | 2.18 (1.16, 4.12) |
| Delavirdina | 400 tres veces al día | 600 tres veces al día, 7 días | 28 | No hay cambios significativos | 1.53 (1.07, 2.20) |
3.98 * (2.04, 7.78) |
| Efavirenz † | 600 una vez al día, 10 días | 1000 tres veces al día, 10 días | 20 | |||
| Después de la dosis de la mañana | Sin cambios significativos * | 0,67 * (0.61, 0.74) |
0.61 * (0.49, 0.76) |
|||
| Después de la dosis de la tarde | Sin cambios significativos * | 0,63 * (0.54, 0.74) |
0,48 * (0.43, 0.53) |
|||
| Después de la dosis de la tarde | 0,71 * (0.57, 0.89) |
0,54 * (0.46, 0.63) |
0,43 * (0.37, 0.50) |
|||
| Fluconazol † | 400 una vez al día, 8 días | 1000 tres veces al día, 7 días | 11 | 0,87 (0.72, 1.05) |
0,76 (0.59, 0.98) |
0,90 (0.72, 1.12) |
| Jugo de pomelo | 8 oz. | 400 dosis única | 10 | 0.65 (0.53, 0.79) |
0,73 (0.60, 0.87) |
0,90 (0.71, 1.15) |
| Isoniazid | 300 una vez al día por la mañana, 8 días | 800 tres veces al día, 7 días | 11 | 0.95 (0.88, 1.03) |
0.99 (0.87, 1.13) |
0,89 (0.75, 1.06) |
| Itraconazol | 200 dos veces al día, 7 días | 600 tres veces al día, 7 días | 12 | 0,78 (0.69, 0.88) |
0,99 * (0.91, 1.06) |
1.49 (1.28, 1.74) |
| Ketoconazol | 400 una vez al día, 7 días | 600 tres veces al día, 7 días | 12 | 0,69 * (0.61, 0.78) |
0,80 * (0.74, 0.87) |
1.29 (1.11, 1.51) |
| 400 una vez al día, 7 días | 400 tres veces al día, 7 días | 12 | 0,42 * (0.37, 0.47) |
0,44 * (0.41, 0.48) |
0,73 * (0.62, 0.85) |
|
| Metadona | 20-60 una vez al día por la mañana, 8 días | 800 tres veces al día, 8 días | 10 | Vea el texto a continuación para discutir la interacción. | ||
| Quinidina | 200 dosis única | 400 dosis única | 10 | 0.96 (0.79, 1.18) |
1.07 (0.89, 1.28) |
0.93 (0.73, 1.19) |
| Rifabutina | 150 una vez al día por la mañana, 10 días | 800 tres veces al día, 10 días | 14 | 0,80 (0.72, 0.89) |
0.68 (0.60, 0.76) |
0.60 (0.51, 0.72) |
| Rifabutina | 300 una vez al día por la mañana, 10 días | 800 tres veces al día, 10 días | 10 | 0,75 (0.61, 0.91) |
0.66 (0.56, 0.77) |
0.61 (0.50, 0.75) |
| Rifampina | 600 una vez al día por la mañana, 8 días | 800 tres veces al día, 7 días | 12 | 0.13 (0.08, 0.22) |
0.08 (0.06, 0.11) |
No hecho |
| Ritonavir | 100 dos veces al día, 14 días | 800 dos veces al día, 14 días | 10, 16 ‡ | Vea el texto a continuación para discutir la interacción. | ||
| Ritonavir | 200 dos veces al día, 14 días | 800 dos veces al día, 14 días | 9, 16 ‡ | Vea el texto a continuación para discutir la interacción. | ||
| Sildenafil | 25 dosis única | 800 tres veces al día | 6 | Vea el texto a continuación para discutir la interacción. | ||
| S t. Hierba de John (Hypericum perforatum, estandarizada a 0.3% de hipericina) | 300 tres veces al día con comidas, 14 días | 800 tres veces al día | 8 | No disponible | 0,46 (0.34, 0.58) § |
0.19 (0.06, 0.33) § |
| Stavudine (d4T) † |
40 dos veces al día, 7 días | 800 tres veces al día, 7 días | 11 | 0.95 (0.80, 1.11) |
0.95 (0.80, 1.12) |
1.13 (0.83, 1.53) |
| Trimetoprima / sulfametoxazol | 800 trimetoprima / 160 sulfametoxazol q12h, 7 días | 400 cuatro veces al día, 7 días | 12 | 1.12 (0.87, 1.46) |
0,98 (0.81, 1.18) |
0,83 (0.72, 0.95) |
| Zidovudina † | 200 tres veces al día, 7 días | 1000 tres veces al día, 7 días | 12 | 1.06 (0.91, 1.25) |
1.05 (0.86, 1.28) |
1.02 (0.77, 1.35) |
| Zidovudina / Lamivudina (3TC) † |
200/150 tres veces al día, 7 días | 800 tres veces al día, 7 días | 6, 9¶ | 1.05 (0.83, 1.33) |
1.04 (0.67, 1.61) |
0,98 (0.56, 1.73) |
| Todos los estudios de interacción realizados en saludable
Sujetos adultos VIH negativos, a menos que se indique lo contrario. * Relativo a indinavir 800 mg tres veces al día solo. † Estudio realizado en sujetos VIH positivos. ‡ Comparación con datos históricos sobre 16 sujetos que reciben indinavir solo. § 95% CI . ¶ Diseño de grupo paralelo; n para indinavir + fármaco coadministrado, n para indinavir solo. |
Tabla 3: Interacciones farmacológicas: parámetros farmacocinéticos
para el fármaco coadministrado en presencia de indinavir (ver PRECAUCIONES, Tabla 9 para
Alteraciones recomendadas en dosis o régimen)
| Droga coadministrada | Dosis de fármaco coadministrado (mg) | Dosis de CRIXIVAN (mg) | n | Relación (con / sin CRIXIVAN) de parámetros farmacocinéticos de medicamentos administrados conjuntamente (90% CI); Sin efecto = 1.00 | ||
| Cmax | AUC | Cmin | ||||
| Claritromicina | 500 dos veces al día, 7 días | 800 tres veces al día, 7 días | 12 | 1.19 (1.02, 1.39) |
1.47 (1.30, 1.65) |
1.97 (1.58, 2.46) n = 11 |
| Efavirenz | 200 una vez al día, 14 días | 800 tres veces al día, 14 días | 20 | No hay cambios significativos | No hay cambios significativos | -- |
| Etinil Estradiol (ORTHO-NOVUM 1/35) * | 35 mcg, 8 días | 800 tres veces al día, 8 días | 18 | 1.02 (0.96, 1.09) |
1.22 (1.15, 1.30) |
1.37 (1.24, 1.51) |
| Isoniazid | 300 una vez al día por la mañana, 8 días | 800 tres veces al día, 8 días | 11 | 1.34 (1.12, 1.60) |
1.12 (1.03, 1.22) |
1.00 (0.92, 1.08) |
| Metadona † | 20-60 una vez al día por la mañana, 8 días | 800 tres veces al día, 8 días | 12 | 0.93 (0.84, 1.03) |
0.96 (0.86, 1.06) |
1.06 (0.94, 1.19) |
| Noretindrona (ORTHO-NOVUM 1/35) * |
1 mcg, 8 días | 800 tres veces al día, 8 días | 18 | 1.05 (0.95, 1.16) |
1.26 (1.20, 1.31) |
1.44 (1.32, 1.57) |
| Rifabutina 150 mg una vez al día por la mañana, 11 días + indinavir en comparación con 300 mg una vez al día por la mañana, 11 días solo | 150 una vez al día por la mañana, 10 días | 800 tres veces al día, 10 días | 14 | 1.29 (1.05, 1.59) |
1.54 (1.33, 1.79) |
1.99 (1.71, 2.31) n = 13 |
| 300 una vez al día por la mañana, 10 días | 800 tres veces al día, 10 días | 10) | 2.34 (1.64, 3.35) |
2.73 (1.99, 3.77) |
3.44 (2.65, 4.46) n = 9 |
|
| Ritonavir | 100 dos veces al día, 14 días | 800 dos veces al día, 14 días | 10, 4 ‡ | 1.61 (1.13, 2.29) |
1.72 (1.20, 2.48) |
1.62 (0.93, 2.85) |
| 200 dos veces al día, 14 días | 800 dos veces al día, 14 días | 9, 5 ‡ | 1.19 (0.85, 1.66) |
1.96 (1.39, 2.76) |
4.71 (2.66, 8.33) n = 9, 4 |
|
| Saquinavir | ||||||
| Formulación de gel duro | 600 dosis única | 800 tres veces al día, 2 días | 6 | 4.7 (2.7, 8.1) |
6.0 (4.0, 9.1) |
2.9 § (1.7, 4.7) |
| Formulación de gel suave Formulación de gel suave | 800 dosis única 1200 dosis única | 800 tres veces al día, 2 días 800 tres veces al día, 2 días | 6 | 6.5 (4.7, 9.1) |
7.2 (4.3, 11.9) |
5.5 § (2.2, 14.1) |
| 6 | 4.0 (2.7, 5.9) |
4.6 (3.2, 6.7) |
5.5 § (3.7, 8.3) |
|||
| Sildenafil | 25 dosis única | 800 tres veces al día | 6 | Vea el texto a continuación para discutir la interacción. | ||
| Stavudine¶ | 40 dos veces al día, 7 días | 800 tres veces al día, 7 días | 13 | 0,86 (0.73, 1.03) |
1.21 (1.09, 1.33) |
No hecho |
| Teofilina | 250 dosis única (en los días 1 y 7) | 800 tres veces al día, 6 días (Días 2 a 7) |
12, 4 ‡ | 0,88 (0.76, 1.03) |
1.14 (1.04, 1.24) |
1.13 (0.86, 1.49) n = 7, 3 |
| Trimetoprima / sulfametoxazol | ||||||
| Trimetoprima | 800 trimetoprima / 160 sulfametoxazol q12h, 7 días | 400 q6h, 7 días | 12 | 1.18 (1.05, 1.32) |
1.18 (1.05, 1.33) |
1.18 (1.00, 1.39) |
| Trimetoprima / sulfametoxazol | ||||||
| Sulfametoxazol | 800 trimetoprima / 160 sulfametoxazol q12h, 7 días | 400 q6h, 7 días | 12 | 1.01 (0.95, 1.08) |
1.05 (1.01, 1.09) |
1.05 (0.97, 1.14) |
| Vardenafil | 10 dosis única | 800 tres veces al día | 18 | Vea el texto a continuación para discutir la interacción. | ||
| Zidovudina¶ | 200 tres veces al día, 7 días | 1000 tres veces al día, 7 días | 12 | 0,89 (0.73, 1.09) |
1.17 (1.07, 1.29) |
1.51 (0.71, 3.20) n = 4 |
| Zidovudina / Lamivudina¶ | ||||||
| Zidovudina | 200/150 tres veces al día, 7 días | 800 tres veces al día, 7 días | 6, 7 ‡ | 1.23 (0.74, 2.03) |
1.39 (1.02, 1.89) |
1.08 (0.77, 1.50) n = 5, 5 |
| Zidovudina / Lamivudina¶ | ||||||
| Lamivudina | 200/150 tres veces al día, 7 días | 800 tres veces al día, 7 días | 6, 7 ‡ | 0,73 (0.52, 1.02) |
0.91 (0.66, 1.26) |
0,88 (0.59, 1.33) |
| Todos los estudios de interacción realizados en saludable
Sujetos adultos VIH negativos, a menos que se indique lo contrario. * Marca registrada de Ortho Pharmaceutical Corporation. † Estudio realizado en sujetos sobre mantenimiento con metadona. ‡ Diseño de grupo paralelo; n para fármaco coadministrado + indinavir, n para droga coadministrada sola. § C6hr ¶ Estudio realizado en sujetos VIH positivos. |
Delavirdina
La delavirdina inhibe el metabolismo de indinavir esa administración conjunta de indinavir de 400 mg o 600 mg tres veces al día con La delavirdina de 400 mg tres veces al día altera el AUC, la Cmáx y la Cmin de indinavir (ver tabla 2). Indinavir no tuvo efecto sobre la farmacocinética de delavirdina (ver DOSIS Y ADMINISTRACIÓN, Terapia concomitante, Delavirdina), basado en una comparación con datos farmacocinéticos históricos de delavirdina.
Metadona
Administración de indinavir (800 mg cada 8 horas) con metadona (20 mg a 60 mg diarios) durante una semana en sujetos con metadona el mantenimiento no resultó en cambios en el AUC de metadona. Basado en una comparación con datos históricos, hubo poco o ningún cambio en el AUC de indinavir
Ritonavir
En comparación con los datos históricos en pacientes que recibieron indinavir 800 mg cada 8 horas solo, administración conjunta dos veces al día resultaron voluntarios de indinavir 800 mg y ritonavir con alimentos durante dos semanas en un aumento de 2.7 veces del AUC24h de indinavir, un aumento de 1.6 veces en la Cmáx de indinavir, y un aumento de 11 veces en indinavir Cmin para una dosis de ritonavir de 100 mg y a Aumento de 3.6 veces del indinavir AUC24h, un aumento de 1.8 veces en la Cmáx de indinavir y un aumento de 24 veces en indinavir Cmin para una dosis de ritonavir de 200 mg. En el mismo estudio, administración conjunta dos veces al día de indinavir (800 mg) y ritonavir (100 o 200 mg) dio como resultado aumentos de ritonavir AUC24h versus las mismas dosis de ritonavir solo (ver Tabla 3).
Sildenafil
Los resultados de un estudio publicado en hombres infectados por el VIH (n = 6) indicó que la administración conjunta de indinavir (800 mg cada 8 horas crónicamente) con una dosis única de 25 mg de sildenafil resultó en un aumento del 11% en promedio AUC0-8hr de indinavir y un aumento del 48% en el pico promedio de indinavir concentración (Cmáx) en comparación con 800 mg cada 8 horas solo. Sildenafil promedio El AUC aumentó en un 340% después de la administración conjunta de sildenafil y indinavir en comparación con los datos históricos después de la administración de sildenafil solo (ver CONTRAINDICACIONES, ADVERTENCIAS, INTERACCIONES DE DROGAS y PRECAUCIONES : INTERACCIONES DE DROGAS).
Vardenafil
Indinavir (800 mg cada 8 horas) coadministrado con a una dosis única de 10 mg de vardenafil resultó en un aumento de 16 veces en el vardenafil AUC, un aumento de 7 veces en la Cmáx de vardenafil y un aumento de 2 veces en el vardenafil vida media (ver ADVERTENCIAS, INTERACCIONES DE DROGAS y PRECAUCIONES : INTERACCIONES DE DROGAS).
Embarazo Categoría C: Toxicidad del desarrollo Se realizaron estudios en conejos (a dosis de hasta 240 mg / kg / día), perros (a dosis de hasta 80 mg / kg / día) y ratas (a dosis de hasta 640 mg / kg / día). El más alto Las dosis en estos estudios produjeron exposiciones sistémicas en estas especies comparables o ligeramente mayor que la exposición humana. No hay tratamiento externo relacionado Se observaron cambios viscerales o esqueléticos en conejos o perros. No Se observaron cambios externos o viscerales relacionados con el tratamiento en ratas. Aumentos relacionados con el tratamiento sobre los controles en la incidencia de costillas supernumerarias (a exposiciones iguales o inferiores a las de los humanos) y de costillas cervicales (a exposiciones comparable o ligeramente mayor que los de los humanos) se observaron en ratas. En las tres especies, sin efectos relacionados con el tratamiento sobre la supervivencia embrionaria / fetal o se observaron pesos fetales.
En conejos, a una dosis materna de 240 mg / kg / día, sin drogas se detectó en plasma fetal 1 hora después de la dosificación. Niveles de fármaco en plasma fetal 2 horas después de la dosificación fueron aproximadamente el 3% de los niveles de drogas en plasma materno. En perros, a una dosis materna de 80 mg / kg / día, los niveles de fármaco en plasma fetal fueron aproximadamente el 50% de los niveles de drogas en plasma materno, tanto 1 como 2 horas después dosificación. En ratas, a dosis maternas de 40 y 640 mg / kg / día, fármaco plasmático fetal los niveles fueron aproximadamente del 10 al 15% y del 10 al 20% de la droga plasmática materna niveles 1 y 2 horas después de la dosificación, respectivamente.
Indinavir se administró a los monos Rhesus durante el tercer trimestre del embarazo (a dosis de hasta 160 mg / kg dos veces al día) y hasta monos Rhesus neonatales (a dosis de hasta 160 mg / kg dos veces al día). Cuando se administra a los recién nacidos, indinavir causó una exacerbación de la hiperbilirrubinemia fisiológica transitoria visto en esta especie después del nacimiento; Los valores séricos de bilirrubina fueron aproximadamente cuatro veces por encima de los controles a 160 mg / kg dos veces al día. Una exacerbación similar lo hizo no ocurre en neonatos después de la exposición en el útero a indinavir durante el tercero trimestre del embarazo. En los monos Rhesus, los niveles de drogas en plasma fetal fueron aproximadamente del 1 al 2% de los niveles de drogas en plasma materno aproximadamente 1 hora después dosificación materna a 40, 80 o 160 mg / kg dos veces al día.
Se ha producido hiperbilirrubinemia durante el tratamiento con CRIXIVAN (ver PRECAUCIONES y REACCIONES ADVERSAS). Se desconoce si CRIXIVAN administrado a la madre en el período perinatal exacerbará hiperbilirrubinemia fisiológica en neonatos.
No hay estudios adecuados y bien controlados en pacientes embarazadas. CRIXIVAN debe usarse durante el embarazo solo si el El beneficio potencial justifica el riesgo potencial para el feto.
Una dosis de CRIXIVAN de 800 mg cada 8 horas (con zidovudina Se han estudiado 200 mg cada 8 horas y lamivudina 150 mg dos veces al día en 16 infectados por el VIH pacientes embarazadas a las 14 a 28 semanas de gestación al momento de la inscripción (estudio PACTG 358). Dadas las exposiciones anteparto sustancialmente más bajas observadas y las limitadas datos en esta población de pacientes, no se recomienda el uso de indinavir en Pacientes embarazadas infectados por el VIH (ver FARMACOLOGÍA CLÍNICA, Embarazada Pacientes).
Registro de embarazo antirretroviral
Monitorear los resultados materno-fetales de las pacientes embarazadas expuesto a CRIXIVAN, se ha establecido un Registro de embarazo antirretroviral. Se alienta a los médicos a registrar pacientes llamando al 1-800-258-4263.
Ensayos clínicos en adultos
Nefrolitiasis / urolitiasis, incluido el dolor en el flanco o sin hematuria (incluida la hematuria microscópica), se ha informado en aproximadamente 12.4% (301/2429; rango en ensayos individuales: 4.7% a 34.4%) de pacientes que reciben CRIXIVAN a la dosis recomendada en ensayos clínicos con una mediana de seguimiento de 47 semanas (rango: 1 día a 242 semanas; 2238 pacientes-años seguimiento). La frecuencia acumulada de eventos de nefrolitiasis aumenta con duración de la exposición a CRIXIVAN; sin embargo, el riesgo a lo largo del tiempo permanece relativamente constante. De los pacientes tratados con CRIXIVAN que se desarrollaron nefrolitiasis / urolitiasis en ensayos clínicos durante la fase doble ciego Se informó que el 2.8% (7/246) desarrolló hidronefrosis y el 4.5% (11/246) se sometió a colocación de stent. Después del episodio agudo, 4.9% (12/246) de los pacientes interrumpieron la terapia. (Ver ADVERTENCIAS y DOSIS Y ADMINISTRACIÓN, Nefrolitiasis / Urolitiasis.)
Hiperbilirrubinemia asintomática (bilirrubina total ≥ 2.5 mg / dL), reportado predominantemente como bilirrubina indirecta elevada, tiene ocurrió en aproximadamente el 14% de los pacientes tratados con CRIXIVAN. En <1% esto se asoció con elevaciones en ALT o AST .
Hiperbilirrubinemia y nefrolitiasis / urolitiasis ocurrió con mayor frecuencia a dosis superiores a 2.4 g / día en comparación con las dosis ≤ 2.4 g / día.
Experiencias adversas clínicas informadas en ≥ 2% de pacientes tratados con CRIXIVAN solo, CRIXIVAN en combinación con zidovudina o zidovudina más lamivudina, zidovudina sola o zidovudina más lamivudina se presentan en la Tabla 10.
Tabla 10: Experiencias clínicas adversas informadas en ≥ 2%
de pacientes
| Experiencia adversa | Estudio 028 Considerado relacionado con drogas y de intensidad moderada o severa | Estudie ACTG 320 de Relación desconocida con medicamentos y de Intensidad severa o potencialmente mortal | |||
| CRIXIVAN Porcentaje (n = 332) |
CRIXIVAN más Zidovudine Percent (n = 332) |
Porcentaje de zidovudina (n = 332) |
CRIXIVAN más Zidovudina más Lamivudina por ciento (n = 571) |
Zidovudina más Lamivudina Porcentaje (n = 575) |
|
| El cuerpo como un todo | |||||
| Dolor abdominal | 16,6 | 16.0 | 12,0 | 1.9 | 0.7 |
| Astenia / fatiga | 2.1 | 4.2 | 3.6 | 2.4 | 4.5 |
| Fiebre | 1.5 | 1.5 | 2.1 | 3.8 | 3.0 |
| Malestar | 2.1 | 2.7 | 1.8 | 0 | 0 |
| Sistema digestivo | |||||
| Náuseas | 11,7 | 31,9 | 19.6 | 2.8 | 1.4 |
| Diarrea | 3.3 | 3.0 | 2.4 | 0.9 | 1.2 |
| Vómitos | 8.4 | 17.8 | 9.0 | 1.4 | 1.4 |
| Regurgitación ácida | 2.7 | 5.4 | 1.8 | 0.4 | 0 |
| Anorexia | 2.7 | 5.4 | 3.0 | 0.5 | 0.2 |
| Aumento de apetito | 2.1 | 1.5 | 1.2 | 0 | 0 |
| Dispepsia | 1.5 | 2.7 | 0.9 | 0 | 0 |
| Ictericia | 1.5 | 2.1 | 0.3 | 0 | 0 |
| Sistema Hemico y Linfático | |||||
| Anemia | 0.6 | 1.2 | 2.1 | 2.4 | 3.5 |
| Sistema musculoesquelético | |||||
| Dolor de espalda | 8.4 | 4.5 | 1.5 | 0.9 | 0.7 |
| Sistema nervioso / psiquiátrico | |||||
| Dolor de cabeza | 5.4 | 9.6 | 6.0 | 2.4 | 2.8 |
| Mareo | 3.0 | 3.9 | 0.9 | 0.5 | 0.7 |
| Somnolencia | 2.4 | 3.3 | 3.3 | 0 | 0 |
| Apéndice de piel y piel | |||||
| Prurito | 4.2 | 2.4 | 1.8 | 0.5 | 0 |
| Sarpullido | 1.2 | 0.6 | 2.4 | 1.1 | 0.5 |
| Sistema respiratorio | |||||
| Tos | 1.5 | 0.3 | 0.6 | 1.6 | 1.0 |
| Dificultad para respirar / disnea / falta de aliento | 0 | 0.6 | 0.3 | 1.8 | 1.0 |
| Sistema urogenital | |||||
| Nefrolitiasis / urolitiasis | 8.7 | 7.8 | 2.1 | 2.6 | 0.3 |
| Disuria | 1.5 | 2.4 | 0.3 | 0.4 | 0.2 |
| Sentidos especiales | |||||
| Perversión de sabor | 2.7 | 8.4 | 1.2 | 0.2 | 0 |
| * Incluyendo cólico renal, y dolor en el flanco con y sin hematuria | |||||
En las fases I y II controladas ensayos, los siguientes eventos adversos se informaron significativamente más frecuentemente por aquellos asignados al azar a los brazos que contienen CRIXIVAN que por aquellos aleatorizado a análogos de nucleósidos: erupción cutánea, infección de las vías respiratorias superiores, seco piel, faringitis, perversión del gusto.
Laboratorio seleccionado anormalidades de intensidad severa o potencialmente mortal reportadas en pacientes tratado con CRIXIVAN solo, CRIXIVAN en combinación con zidovudina o zidovudina más lamivudina, zidovudina sola o zidovudina más lamivudina son presentado en la Tabla 11.
Tabla 11: Anomalías de laboratorio seleccionadas de graves
o Intensidad potencialmente mortal informada en los estudios 028 y ACTG 320
| Estudio 028 Considerado relacionado con drogas y de intensidad moderada o severa | Estudie ACTG 320 de Relación desconocida con medicamentos y de Intensidad severa o potencialmente mortal | ||||
| CRIXIVAN Porcentaje (n = 329) |
CRIXIVAN más Zidovudine Percent (n = 320) |
Porcentaje de zidovudina (n = 330) |
CRIXIVAN más Zidovudina más Lamivudina por ciento (n = 571) |
Zidovudina más Lamivudina Porcentaje (n = 575) |
|
| Hematología | |||||
| Hemoglobina disminuida <7.0 g / dL | 0.6 | 0.9 | 3.3 | 2.4 | 3.5 |
| Recuento de plaquetas disminuido <50 THS / mm³ | 0.9 | 0.9 | 1.8 | 0.2 | 0.9 |
| Disminución de neutrófilos <0.75 THS / mm³ | 2.4 | 2.2 | 6.7 | 5.1 | 14,6 |
| Química de la sangre | |||||
| Aumento de ALT> 500% ULN * | 4.9 | 4.1 | 3.0 | 2.6 | 2.6 |
| Aumento de AST> 500% ULN | 3.7 | 2.8 | 2.7 | 3.3 | 2.8 |
| Bilirrubina sérica total> 250% ULN | 11,9 | 9.7 | 0.6 | 6.1 | 1.4 |
| Aumento de la amilasa sérica> 200% ULN | 2.1 | 1.9 | 1.8 | 0.9 | 0.3 |
| Aumento de glucosa> 250 mg / dL | 0.9 | 0.9 | 0.6 | 1.6 | 1.9 |
| Aumento de creatinina> 300% ULN | 0 | 0 | 0.6 | 0.2 | 0 |
| * Límite superior de lo normal rango. | |||||
Experiencia posterior a la comercialización
Cuerpo en su conjunto: redistribución / acumulación de grasa corporal (ver PRECAUCIONES, Redistribución gorda).
Sistema cardiovascular: trastornos cardiovasculares incluyendo infarto de miocardio y angina de pecho; trastorno cerebrovascular.
Sistema digestivo: función hepática anormalidades; hepatitis incluyendo informes de insuficiencia hepática (ver ADVERTENCIAS); pancreatitis ictericia distensión abdominal dispepsia.
Hematológico: aumento espontáneo sangrado en pacientes con hemofilia (ver PRECAUCIONES); hemolítico agudo anemia (ver ADVERTENCIAS).
Endocrino / Metabólico: diabetes de nueva aparición mellitus, exacerbación de diabetes mellitus preexistente, hiperglucemia (ver ADVERTENCIAS).
Hipersensibilidad : reacciones anafilactoides; urticaria vasculitis.
Sistema musculoesquelético : artralgia, periartritis.
Sistema nervioso / psiquiátrico : parestesia oral; depresión.
Apéndice de piel y piel: erupción cutánea, incluido eritema multiforme y síndrome de Stevens-Johnson; hiperpigmentación; alopecia encarnado uñas de los pies y / o paroniquia; prurito.
Sistema urogenital : nefrolitiasis / urolitiasis, en algunos casos que resultan en insuficiencia renal o insuficiencia renal aguda, pielonefritis con o sin bacteriemia (ver ADVERTENCIAS); intersticial nefritis a veces con depósitos de cristal de indinavir; en algunos pacientes, el la nefritis intersticial no se resolvió después de la interrupción de CRIXIVAN ; insuficiencia renal; insuficiencia renal; leucocituria (ver PRECAUCIONES), cristaluria disuria.
Anomalías de laboratorio
Aumento de triglicéridos séricos; aumento del colesterol sérico.
Ha habido más de 60 informes de agudos o crónicos sobredosis humana (hasta 23 veces la dosis diaria total recomendada de 2400 mg) con CRIXIVAN. Los síntomas más comúnmente reportados fueron renales (p. Ej., nefrolitiasis / urolitiasis, dolor en el flanco, hematuria) y gastrointestinal (p. ej., náuseas, vómitos, diarrea).
No se sabe si CRIXIVAN es dializable por peritoneal o hemodiálisis.
Absorción
Indinavir se absorbió rápidamente en ayunas con a tiempo hasta la concentración plasmática máxima (Tmax) de 0.8 ± 0.3 horas (media ± S.D.) (n = 11). Un aumento mayor que la dosis proporcional en el plasma de indinavir se observaron concentraciones en el rango de dosis de 200-1000 mg. A una dosificación régimen de 800 mg cada 8 horas, área de estado estacionario debajo del plasma la curva de tiempo de concentración (AUC) fue de 30,691 ± 11,407 nMÃ ¢ â ¬ ¢ hora (n = 16), plasma máximo la concentración (Cmáx) fue de 12,617 ± 4037 nM (n = 16) y la concentración plasmática ocho horas después de la dosis (trasero) fue de 251 ± 178 nM (n = 16).
Efecto de los alimentos sobre la absorción oral
Administración de indinavir con una comida rica en calorías grasa y proteína (784 kcal, 48,6 g de grasa, 31,3 g de proteína) dieron como resultado un 77% ± 8% reducción en el AUC y una reducción del 84% ± 7% en la Cmáx (n = 10). Administración con comidas más ligeras (p. ej., una comida de tostadas secas con gelatina, jugo de manzana y café con leche descremada y azúcar o una comida de copos de maíz, leche descremada y azúcar) resultó en poco o ningún cambio en el AUC, la Cmáx o la concentración mínima.
Distribución
Indinavir se unió aproximadamente en un 60% al plasma humano proteínas en un rango de concentración de 81 nM a 16.300 nM
Metabolismo
Después de una dosis de 400 mg de 14C-indinavir, 83 ± 1% (n = 4) y 19 ± 3% (n = 6) de la radiactividad total se recuperó en heces y orina respectivamente; la radiactividad debida al fármaco original en heces y orina fue del 19,1% y 9.4%, respectivamente. Se han identificado siete metabolitos, un conjugado de glucurónido y seis metabolitos oxidativos. In vitro los estudios indican que el citocromo P-450 3A4 (CYP3A4) es la enzima principal responsable de la formación del oxidativo metabolitos.
Eliminación
Menos del 20% de indinavir se excreta sin cambios en el orina. La excreción urinaria media del fármaco inalterado fue de 10,4 ± 4,9% (n = 10) y 12,0 ± 4.9% (n = 10) después de una dosis única de 700 mg y 1000 mg, respectivamente. Indinavir se eliminó rápidamente con una vida media de 1.8 ± 0.4 horas (n = 10). No se observó una acumulación significativa después de dosis múltiples a 800 mg cada una 8 horas.
Disponible en países
 Australia
Australia Austria
Austria Belgium
Belgium Brazil
Brazil Bulgaria
Bulgaria Canada
Canada China
China Cyprus
Cyprus Czech Republic
Czech Republic Denmark
Denmark Estonia
Estonia Finland
Finland France
France Germany
Germany Greece
Greece Hong Kong
Hong Kong Hungary
Hungary Iceland
Iceland Ireland
Ireland Italy
Italy Japan
Japan Latvia
Latvia Lebanon
Lebanon Liechtenstein
Liechtenstein Lithuania
Lithuania Luxembourg
Luxembourg Malta
Malta Netherlands
Netherlands New Zealand
New Zealand Norway
Norway Philippines
Philippines Poland
Poland Portugal
Portugal Romania
Romania Russia
Russia Serbia
Serbia Slovakia
Slovakia Slovenia
Slovenia South Africa
South Africa Spain
Spain Sweden
Sweden Switzerland
Switzerland Taiwan
Taiwan Turkey
Turkey United Kingdom
United Kingdom USA
USA Venezuela
Venezuela