

Componentes:
Método de ação:
Opção de tratamento:
Medicamente revisado por Fedorchenko Olga Valeryevna, Farmácia Última atualização em 26.06.2023

Atenção! As informações na página são apenas para profissionais de saúde! As informações são coletadas em fontes abertas e podem conter erros significativos! Tenha cuidado e verifique novamente todas as informações desta página!
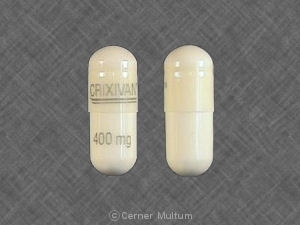
Cápsulas de CRIXIVAN são fornecidos da seguinte forma:
No Cápsulas de 3756 - 200 mg: branco semi-translúcido. cápsulas codificadas “CRIXIVAN ™ 200 mg” em azul.
Disponível como:
NDC 0006-0571-43 frascos de unidade de uso de 360 (com dessecante).
No Cápsulas de 3758 - 400 mg: branco semi-translúcido. cápsulas codificadas “CRIXIVAN ™ 400 mg” em verde. Disponível como:
NDC 0006-0573-62 frascos de unidade de uso de 180 (com dessecante)
Armazenamento
Garrafas: Armazene em um recipiente bem fechado em temperatura ambiente, 15-30 ° C (59-86 ° F). Proteger da umidade.
As cápsulas de CRIXIVAN são sensíveis à umidade. CRIXIVAN deve ser dispensado e armazenado no recipiente original. O dessecante deveria permaneça na garrafa original.
Dist. por: Merck Sharp & Dohme Corp., uma subsidiária da MERCK & CO., INC., Whitehouse Station, NJ 08889, EUA. Revisado em 03/2015
CRIXIVAN em combinação com agentes anti-retrovirais é indicado para o tratamento da infecção pelo HIV. Esta indicação é baseada em dois ensaios clínicos de aproximadamente 1 ano de duração que demonstraram: 1) uma redução no risco de doenças ou morte definidoras de AIDS; 2) uma supressão prolongada de RNA do HIV .
A dose recomendada de CRIXIVAN é de 800 mg (geralmente dois Cápsulas de 400 mg) por via oral a cada 8 horas.
CRIXIVAN deve ser tomado em intervalos de 8 horas. Para absorção ideal, CRIXIVAN deve ser administrado sem alimentos, mas com água 1 hora antes ou 2 horas após uma refeição. Alternativamente, CRIXIVAN pode ser administrado com outros líquidos, como leite desnatado, suco, café ou chá, ou com uma refeição leve, p., torradas secas com geléia, suco e café com leite desnatado e açúcar; ou flocos de milho, leite desnatado e açúcar. (Vejo FARMACOLOGIA CLÍNICA, Efeito dos alimentos na absorção oral.)
Para garantir uma hidratação adequada, recomenda-se isso adultos bebem pelo menos 1,5 litros (aproximadamente 48 onças) de líquidos durante o curso de 24 horas.
Terapia concomitante (Vejo FARMACOLOGIA CLÍNICA, Interações medicamentosase / ou PRECAUÇÕES: INTERAÇÕES DE DROGAS.)
Delavirdine
A redução da dose de CRIXIVAN para 600 mg a cada 8 horas deve ser considerado ao administrar delavirdina 400 mg três vezes ao dia.
Didanosina
Se indinavir e didanosina são administrados concomitantemente, eles devem ser administrados com pelo menos uma hora de intervalo em um vazio estômago (consulte a circular do produto do fabricante para didanosina).
Itraconazol
A redução da dose de CRIXIVAN para 600 mg a cada 8 horas é recomendado ao administrar itraconazol 200 mg duas vezes ao dia simultaneamente.
Cetoconazol
A redução da dose de CRIXIVAN para 600 mg a cada 8 horas é recomendado ao administrar cetoconazol simultaneamente.
Rifabutin
Redução da dose de rifabutina para metade da dose padrão (consulte a circular do produto do fabricante para rifabutina) e um aumento da dose CRIXIVAN a 1000 mg a cada 8 horas é recomendado quando a rifabutina e CRIXIVAN são co-administrados.
Insuficiência hepática
A dose de CRIXIVAN deve ser reduzida para 600 mg a cada 8 horas em pacientes com insuficiência hepática leve a tomoderado devido a cirrose.
Nefrolitíase / Urolitíase
Além da hidratação adequada, o gerenciamento médico está dentro pacientes que apresentam nefrolitíase / urolitíase podem incluir temporário interrupção (por exemplo,., 1 a 3 dias) ou descontinuação da terapia.
CRIXIVAN está contra-indicado em pacientes com clinicamente hipersensibilidade significativa a qualquer um de seus componentes.
A inibição do CYP3A4 pelo CRIXIVAN pode resultar em elevada concentrações plasmáticas dos seguintes medicamentos, potencialmente causando graves ou reações com risco de vida:
Tabela 7: Interações medicamentosas com Crixivan :
Drogas contra-indicadas
| Classe de drogas | Drogas dentro da classe que são contra-indicadas com o CRIXIVAN |
| Antagonista alfa do receptor 1 | alfuzosina |
| Antiarrítmicos | amiodarona |
| Derivados de ergot | di-hidroergotamina, ergonovina, ergotamina, metilergonovina |
| Agentes de motilidade gastrointestinal | cisaprida |
| Inibidores da HMG-CoA Reductase | lovastatina, sinvastatina |
| Neurolépticos | pimozida |
| Inibidores do PDE5 | Revatio (sildenafil) [para tratamento da hipertensão arterial pulmonar] |
| Sedativo / hipnótico | midazolam oral, triazolam, alprazolam |
| * Marca registrada da Pfizer, Inc. |
AVISO
ALERTA: Descubra os medicamentos que NÃO deveriam ser tomado com CRIXIVAN . Esta declaração está incluída no frasco do produto etiqueta.
Nefrolitíase / Urolitíase
Nefrolitíase / urolitíase ocorreu com CRIXIVAN terapia. A frequência cumulativa de nefrolitíase é substancialmente mais alta pacientes pediátricos (29%) do que em pacientes adultos (12,4%; variam entre indivíduos ensaios: 4,7% a 34,4%). A frequência cumulativa de eventos de nefrolitíase aumenta com o aumento da exposição ao CRIXIVAN; no entanto, o risco ao longo do tempo permanece relativamente constante. Em alguns casos, a nefrolitíase / urolitíase tem foi associado a insuficiência renal ou insuficiência renal aguda, pielonefrite com ou sem bacteremia. Se sinais ou sintomas de nefrolitíase / urolitíase ocorre (incluindo dor no flanco, com ou sem hematúria ou hematúria microscópica), interrupção temporária (por exemplo,.1-3 dias) ou a descontinuação da terapia pode ser considerada. Hidratação adequada é recomendado em todos os pacientes tratados com CRIXIVAN. (Veja REAÇÕES ADVERSAS e DOSAGEM E ADMINISTRAÇÃO, Nefrolitíase / Urolitíase.)
Anemia hemolítica
Anemia hemolítica aguda, incluindo casos que resultam em morte, foi relatada em pacientes tratados com CRIXIVAN. Uma vez que o diagnóstico é aparentes, devem ser medidas apropriadas para o tratamento da anemia hemolítica instituído, incluindo a descontinuação de CRIXIVAN .
Hepatite
Hepatite, incluindo casos que resultam em insuficiência hepática e a morte foi relatada em pacientes tratados com CRIXIVAN. Porque o a maioria desses pacientes tinha condições médicas confusas e / ou estava recebendo terapia (s) concomitante (s), uma relação causal entre CRIXIVAN e estes eventos não foram estabelecidos.
Hiperglicemia
Novo início de diabetes mellitus, exacerbação de pré-existente diabetes mellitus e hiperglicemia foram relatados durante o pós-comercialização vigilância em pacientes infectados pelo HIV que recebem terapia com inibidores de protease. Alguns pacientes necessitaram de iniciação ou ajuste da dose de insulina ou oral agentes hipoglicêmicos para o tratamento desses eventos. Em alguns casos, diabético ocorreu cetoacidose. Nos pacientes que interromperam a protease terapia com inibidores, hiperglicemia persistiu em alguns casos. Porque esses eventos foram relatados voluntariamente durante a prática clínica, estimativas de frequência não pode ser feito e uma relação causal entre a terapia com inibidores de protease e esses eventos não foram estabelecidos.
Risco de reações adversas graves devido a drogas Interações
Início do CRIXIVAN, um inibidor do CYP3A, em pacientes receber medicamentos metabolizados pelo CYP3A ou iniciar medicamentos metabolizado pelo CYP3A em pacientes que já recebem CRIXIVAN, pode aumentar o plasma concentrações de medicamentos metabolizados pelo CYP3A. Início dos medicamentos que inibir ou induzir o CYP3A pode aumentar ou diminuir as concentrações de CRIXIVAN, respectivamente. Essas interações podem levar a:
- Reações adversas clinicamente significativas, potencialmente levando a eventos graves, com risco de vida ou fatais de maiores exposições de medicamentos concomitantes.
- Reações adversas clinicamente significativas de maior exposições de CRIXIVAN .
- Perda do efeito terapêutico do CRIXIVAN e possível desenvolvimento de resistência.
Consulte a Tabela 9 para obter as etapas para evitar ou gerenciar essas possíveis e interações medicamentosas significativas conhecidas, incluindo recomendações posológicas. Considere o potencial para interações medicamentosas antes e durante o CRIXIVAN terapia; revisar medicamentos concomitantes durante o tratamento com CRIXIVAN; e monitor para as reações adversas associadas aos medicamentos concomitantes.
Uso concomitante de CRIXIVAN com lovastatina ou a sinvastatina é contra-indicada devido a um risco aumentado de miopatia, inclusive rabdomiólise. Deve-se ter cuidado se CRIXIVAN for usado simultaneamente com atorvastatina ou rosuvastatina. Titule a atorvastatina e a rosuvastatina doses cuidadosamente e use a dose mais baixa necessária com CRIXIVAN. (Veja PRECAUÇÕES: INTERAÇÕES DE DROGAS.)
O midazolam é extensamente metabolizado pelo CYP3A4. A administração concomitante de CRIXIVAN com ou sem ritonavir pode causar um grande aumento aumento da concentração desta benzodiazepina. Nenhum estudo de interação medicamentosa foi realizado para a administração concomitante de CRIXIVAN com benzodiazepínicos. Com base em dados de outros inibidores do CYP3A4, concentrações plasmáticas de midazolam espera-se que seja significativamente maior quando o midazolam é administrado por via oral. Portanto, CRIXIVAN não deve ser co-administrado com administração oral midazolam (ver CONTRA-INDICAÇÕES), enquanto cautela deve ser usada administração concomitante de CRIXIVAN e midazolam parenteral. Dados de concomitante o uso de midazolam parenteral com outros inibidores da protease sugere uma possível Aumento de 3-4 vezes nos níveis plasmáticos de midazolam. Se CRIXIVAN com ou sem o ritonavir é co-administrado com midazolam parenteral, deve ser feito em a configuração que garanta monitoramento clínico rigoroso e medicina apropriada manejo em caso de depressão respiratória e / ou sedação prolongada. Dosagem a redução do midazolam deve ser considerada, especialmente se mais de um único é administrada a dose de midazolam.
Deve-se ter cuidado especial ao prescrever sildenafil, tadalafil ou vardenafil em pacientes recebendo indinavir. Espera-se a administração concomitante de CRIXIVAN com esses medicamentos aumentar substancialmente as concentrações plasmáticas de sildenafil, tadalafil e vardenafil e pode resultar em um aumento de eventos adversos, incluindo hipotensão, alterações visuais e priapismo, que foram associadas ao sildenafil tadalafil e vardenafil (ver CONTRA-INDICAÇÕES e PRECAUÇÕES: INTERAÇÕES DE DROGAS e INFORMAÇÃO PATIENTE, e o informações completas de prescrição do fabricante para sildenafil, tadalafil ou vardenafil).
Uso concomitante de CRIXIVAN e St. Erva de John (Hypericum perforatum) ou produtos que contenham St. O mosto de John não é recomendado. Co-administração de CRIXIVAN e St. O mosto de John demonstrou diminuir substancialmente concentrações de indinavir (ver FARMACOLOGIA CLÍNICA, DROGAS INTERAÇÕES) e pode levar à perda da resposta virológica e possível resistência ao CRIXIVAN ou à classe de inibidores da protease.
PRECAUÇÕES
Geral
A hiperbilirrubinemia indireta ocorreu com frequência durante o tratamento com CRIXIVAN e raramente foi associado aumentos nas transaminases séricas (ver também REAÇÕES ADVERSAS, Clínico Julgamentos e Experiência pós-comercialização). Não se sabe se CRIXIVAN exacerbará a hiperbilirrubinemia fisiológica observada nos neonatos. (Vejo Gravidez.)
Nefrite tubulointersticial
Relatos de nefrite tubulointersticial com medular calcificação e atrofia cortical foram observadas em pacientes com leucocitúria grave assintomática (> 100 células / campo de alta potência). Pacientes com leucocitúria grave assintomática deve ser seguida de perto e monitorada frequentemente com urinálise. Uma avaliação mais aprofundada do diagnóstico pode ser justificada e a descontinuação de CRIXIVAN deve ser considerada em todos os pacientes com gravidade leucocitúria.
A síndrome de reconstituição imunológica foi relatada em pacientes tratados com terapia antirretroviral combinada, incluindo CRIXIVAN . Durante a fase inicial do tratamento antirretroviral combinado, pacientes cujos as respostas do sistema imunológico podem desenvolver uma resposta inflamatória a indolente ou infecções oportunistas residuais (como Mycobacterium avium infecção, citomegalovírus, Pneumocystis jirovecii pneumonia [PCP] ou tuberculose), o que pode exigir mais avaliação e tratamento. Autoimune distúrbios (como doença de Graves, polimiosite e síndrome de Guillain-Barré) também foram relatados como ocorrendo no cenário de reconstituição imunológica; Contudo, o tempo para iniciar é mais variável e pode ocorrer muitos meses após o início de tratamento.
Condições coexistentes
Pacientes com hemofilia: Houve relatórios sangramento espontâneo em pacientes com hemofilia A e B tratados inibidores de protease. Em alguns pacientes, foi necessário fator VIII adicional. No em muitos dos casos relatados, o tratamento com inibidores da protease foi continuado ou reiniciado. Uma relação causal entre a terapia com inibidores de protease e estes episódios não foram estabelecidos. (Vejo REAÇÕES ADVERSAS, Pós-Marketing Experiência.)
Pacientes com insuficiência hepática devido a cirrose: Nestes pacientes, a dose de CRIXIVAN deve ser reduzida por causa de metabolismo diminuído do CRIXIVAN (ver DOSAGEM E ADMINISTRAÇÃO). Pacientes com insuficiência renal: pacientes com insuficiência renal não foram estudado.
Redistribuição de gordura
Redistribuição / acumulação de gordura corporal, incluindo central obesidade, aumento de gordura dorsocervical (corcunda de búfalo), perda de periferia desperdício facial, aumento dos seios e "aparência carsóide" foram observado em pacientes recebendo terapia anti-retroviral. O mecanismo e as consequências a longo prazo desses eventos são atualmente desconhecidas. Um causal relacionamento não foi estabelecido.
Informações para pacientes
Uma declaração para pacientes e profissionais de saúde é incluído no rótulo do frasco do produto. ALERTA: Descubra sobre medicamentos isso NÃO deve ser tomado com CRIXIVAN . Uma inserção de pacote de pacientes (PPI) para CRIXIVAN está disponível para informações do paciente.
CRIXIVAN não é uma cura para a infecção pelo HIV-1 e pacientes pode continuar a experimentar doenças associadas à infecção pelo HIV-1, incluindo infecções oportunistas. Os pacientes devem permanecer sob os cuidados de um médico ao usar CRIXIVAN .
Os pacientes devem ser aconselhados a evitar fazer as coisas que puderem espalhar a infecção pelo HIV-1 para outras pessoas.
- Não compartilhe agulhas ou outros equipamentos de injeção.
- Não compartilhe itens pessoais que possam ter sangue ou fluidos corporais neles, como escovas de dentes e lâminas de barbear.
- Não faça nenhum tipo de sexo sem proteção. Sempre pratique sexo seguro usando um preservativo de látex ou poliuretano para diminuir a chance de contato sexual com sêmen, secreções vaginais ou sangue.
- Não amamente. Não sabemos se o CRIXIVAN pode seja passado para o seu bebê no leite materno e se isso pode prejudicar o seu bebê. Além disso, as mães com HIV-1 não devem amamentar porque o HIV-1 pode ser passado o bebê no leite materno.
Os pacientes devem ser aconselhados a permanecer sob os cuidados de a médico ao usar CRIXIVAN e não deve modificar ou interromper o tratamento sem primeiro consultar o médico. Portanto, se uma dose for perdida, pacientes deve tomar a próxima dose no horário programado regularmente e não deve dobrar esta dose. A terapia com CRIXIVAN deve ser iniciada e mantida no dosagem recomendada.
CRIXIVAN pode interagir com alguns medicamentos; portanto, os pacientes devem ser aconselhados a relatar ao médico o uso de qualquer outro medicamentos prescritos, não prescritos ou produtos à base de plantas, particularmente St. John erva.
Para uma absorção ideal, CRIXIVAN deve ser administrado sem comida, mas com água 1 hora antes ou 2 horas após uma refeição. Alternativamente, CRIXIVAN pode ser administrado com outros líquidos, como leite desnatado suco, café ou chá ou com uma refeição leve, p., torradas secas com geléia, suco e café com leite desnatado e açúcar; ou flocos de milho, leite desnatado e açúcar (ver FARMACOLOGIA CLÍNICA, Efeito dos alimentos na absorção oral e DOSAGEM E ADMINISTRAÇÃO). Ingestão de CRIXIVAN com uma refeição rica em calorias, gordura e proteína reduz a absorção de indinavir.
Pacientes recebendo uma fosfodiesterase tipo 5 (PDE5) inibidor (sildenafil, tadalafil ou vardenafil) deve ser avisado de que eles pode estar em um risco aumentado de eventos adversos associados ao inibidor da PDE5, incluindo hipotensão, mudanças visuais e priapismo, e deve relatar prontamente qualquer sintomas para seus médicos (ver CONTRA-INDICAÇÕES e AVISO, DROGAS INTERAÇÕES).
Os pacientes devem ser informados dessa redistribuição ou pode ocorrer acúmulo de gordura corporal em pacientes recebendo terapia anti-retroviral e que a causa e os efeitos a longo prazo na saúde dessas condições não são conhecidos neste momento.
As cápsulas de CRIXIVAN são sensíveis à umidade. Pacientes deve ser informado de que CRIXIVAN deve ser armazenado e usado no original o recipiente e o dessecante devem permanecer na garrafa.
Carcinogênese, Mutagênese, Comprometimento de Fertilidade
Estudos de carcinogenicidade foram realizados em camundongos e ratos. Em camundongos, não foi observada incidência aumentada de qualquer tipo de tumor. O mais alto a dose testada em ratos foi de 640 mg / kg / dia; nesta dose, estatisticamente significante aumento da incidência de adenomas da tireóide foi observado apenas em ratos machos. Nisso dose, a exposição sistêmica diária em ratos foi aproximadamente 1,3 vezes maior que exposição sistêmica diária em humanos. Nenhuma evidência de mutagenicidade ou genotoxicidade foi observado em in vitro testes de mutagênese microbiana (Ames), in vitro alcalino ensaios de eluição para quebra de DNA in vitro e in vivo aberração cromossômica estudos e in vitro ensaios de mutagênese em células de mamíferos. Não relacionado ao tratamento efeitos no acasalamento, fertilidade ou sobrevivência de embriões foram observados em ratos fêmeas e nenhum efeito relacionado ao tratamento no desempenho do acasalamento foi observado em ratos machos em doses que proporcionam exposição sistêmica comparável ou ligeiramente superior a essa com a dose clínica. Além disso, nenhum efeito relacionado ao tratamento foi observado na fecundidade ou fertilidade de fêmeas não tratadas acasaladas com machos tratados.
Gravidez
Categoria de gravidez C: Toxicidade para o desenvolvimento foram realizados estudos em coelhos (em doses de até 240 mg / kg / dia), cães (em doses de até 80 mg / kg / dia) e ratos (em doses de até 640 mg / kg / dia). O mais alto doses nesses estudos produziram exposições sistêmicas nessas espécies comparáveis ou ligeiramente superior à exposição humana. Nenhum externo relacionado ao tratamento alterações viscerais ou esqueléticas foram observadas em coelhos ou cães. Não alterações externas ou viscerais relacionadas ao tratamento foram observadas em ratos. Aumentos relacionados ao tratamento sobre controles na incidência de costelas supranumerárias (em exposições iguais ou inferiores às dos seres humanos) e de costelas cervicais (em exposições comparáveis ou ligeiramente superiores aos dos seres humanos) foram observados em ratos. No todas as três espécies, sem efeitos relacionados ao tratamento na sobrevida embrionária / fetal ou pesos fetais foram observados.
Nos coelhos, na dose materna de 240 mg / kg / dia, nenhum medicamento foi detectado no plasma fetal 1 hora após a administração. Níveis fetais de medicamentos no plasma 2 horas após a administração foram aproximadamente 3% dos níveis maternos de medicamentos plasmáticos. No cães, na dose materna de 80 mg / kg / dia, os níveis plasmáticos fetais foram aproximadamente 50% dos níveis de drogas plasmáticas maternas 1 e 2 horas depois dosagem. Em ratos, em doses maternas de 40 e 640 mg / kg / dia, medicamento plasmático fetal os níveis foram de aproximadamente 10 a 15% e 10 a 20% do medicamento materno para plasma níveis 1 e 2 horas após a administração, respectivamente.
O indinavir foi administrado a macacos Rhesus durante o terceiro trimestre de gravidez (em doses de até 160 mg / kg duas vezes ao dia) e para macacos Rhesus neonatais (em doses de até 160 mg / kg duas vezes ao dia). Quando administrado para os neonatos, o indinavir causou uma exacerbação da hiperbilirrubinemia fisiológica transitória visto nesta espécie após o nascimento; os valores séricos de bilirrubina foram aproximadamente quatro vezes acima dos controles a 160 mg / kg duas vezes ao dia. Uma exacerbação semelhante fez não ocorrer em neonatos após exposição no útero ao indinavir durante o terceiro trimestre de gravidez. Nos macacos Rhesus, os níveis fetais de drogas plasmáticas eram aproximadamente 1 a 2% dos níveis de drogas plasmáticas maternas aproximadamente 1 hora depois dosagem materna a 40, 80 ou 160 mg / kg duas vezes ao dia.
Ocorreu hiperbilirrubinemia durante o tratamento com CRIXIVAN (Vejo PRECAUÇÕES e REAÇÕES ADVERSAS). Não se sabe se CRIXIVAN administrado à mãe no período perinatal irá exacerbar hiperbilirrubinemia fisiológica em neonatos.
Não há estudos adequados e bem controlados pacientes grávidas. CRIXIVAN deve ser utilizado durante a gravidez apenas se o o benefício potencial justifica o risco potencial para o feto.
Uma dose de CRIXIVAN de 800 mg a cada 8 horas (com zidovudina 200 mg a cada 8 horas e lamivudina 150 mg duas vezes ao dia) foi estudado em 16 infectados pelo HIV pacientes grávidas com 14 a 28 semanas de gestação na inscrição (estudo PACTG 358). Dadas as exposições antepartum substancialmente mais baixas observadas e as limitadas dados nesta população de pacientes, o uso de indinavir não é recomendado Pacientes grávidas infectadas pelo HIV (ver FARMACOLOGIA CLÍNICA, Grávida Pacientes).
Registro de Gravidez Anti-retroviral
Monitorar os resultados materno-fetais de pacientes grávidas exposto ao CRIXIVAN, foi estabelecido um Registro de Gravidez Anti-retroviral. Os médicos são incentivados a registrar pacientes ligando para 1-800-258-4263.
Mães de enfermagem
Estudos em ratos lactantes demonstraram isso o indinavir é excretado no leite. Embora não se saiba se CRIXIVAN é excretado no leite humano, existe o potencial de efeitos adversos indinavir em lactentes. As mães devem ser instruídas a interromper enfermagem se eles estiverem recebendo CRIXIVAN. Isso é consistente com o recomendação dos Centros de Serviços de Saúde Pública dos EUA para Controle de Doenças e Prevenção que as mães infectadas pelo HIV não amamentam seus bebês para evitar arriscando a transmissão pós-natal do HIV .
Uso pediátrico
O regime posológico ideal para a utilização de indinavir em pacientes pediátricos não foi estabelecido. Uma dose de 500 mg / m² a cada oito horas foi estudado em estudos não controlados de 70 crianças, de 3 a 18 anos era. Os perfis farmacocinéticos do indinavir nesta dose não foram comparáveis aos perfis previamente observados em adultos que recebem a dose recomendada (ver FARMACOLOGIA CLÍNICA, Pediátrico). Embora a supressão viral fosse observado em algumas das 32 crianças que foram seguidas neste regime 24 semanas, uma taxa substancialmente mais alta de nefrolitíase foi relatada quando comparado aos dados históricos de adultos (ver AVISO, Nefrolitíase / Urolitíase). Médicos que consideram o uso de indinavir em pacientes pediátricos sem outros as opções de inibidores de protease devem estar cientes dos dados limitados disponíveis em essa população e o aumento do risco de nefrolitíase.
Uso geriátrico
Os estudos clínicos de CRIXIVAN não incluíram suficiente número de indivíduos com 65 anos ou mais para determinar se eles respondem diferente dos assuntos mais jovens. Em geral, seleção de dose para idosos o paciente deve ser cauteloso, refletindo a maior frequência de diminuição função hepática, renal ou cardíaca e de doença concomitante ou outra droga terapia.
(veja também CONTRA-INDICAÇÕES, AVISO, PRECAUÇÕES: INTERAÇÕES DE DROGAS) Indinavir é um inibidor da isoforma do citocromo P450 CYP3A4. Co-administração de CRIXIVAN e medicamentos metabolizados principalmente pelo CYP3A4 podem resultar em aumento do plasma concentrações do outro medicamento, que podem aumentar ou prolongar sua efeitos terapêuticos e adversos (ver CONTRA-INDICAÇÕES e AVISO). Baseado em in vitro dados em microssomas hepáticos humanos, o indinavir não inibe CYP1A2, CYP2C9, CYP2E1 e CYP2B6. No entanto, o indinavir pode ser um inibidor fraco do CYP2D6.
O indinavir é metabolizado pelo CYP3A4. Drogas que induzem Espera-se que a atividade do CYP3A4 aumente a depuração do indinavir resultando em concentrações plasmáticas reduzidas de indinavir. Co-administração de CRIXIVAN e outros medicamentos que inibem o CYP3A4 podem diminuir a depuração do indinavir e pode resultar em concentrações plasmáticas aumentadas de indinavir.
Estudos de interação medicamentosa foram realizados com CRIXIVAN e outros medicamentos que provavelmente serão co-administrados e alguns medicamentos comumente usados como sondas para interações farmacocinéticas. Os efeitos da administração concomitante de CRIXIVAN na AUC, Cmax e Cmin estão resumidos na Tabela 2 (efeito de outros medicamentos indinavir) e Tabela 3 (efeito do indinavir em outros medicamentos). Para informação em relação às recomendações clínicas, consulte a Tabela 9 em PRECAUÇÕES.
Tabela 2: Interações medicamentosas: parâmetros farmacocinéticos
para o indinavir na presença do medicamento co-administrado (ver PRECAUÇÕES, tabela
9 para alterações recomendadas em dose ou regime)
| Medicamento co-administrado | Dose do medicamento co-administrado (mg) | Dose de CRIXIVAN (mg) | n | Proporção (com / sem medicamento co-administrado) dos parâmetros farmacocinéticos do indinavir (IC 90%); Sem efeito = 1,00 |
||
| Cmax | AUC | Cmin | ||||
| Cimetidina | 600 duas vezes ao dia, 6 dias | 400 dose única | 12 | 1.07 (0,77, 1,49) |
0,98 (0,81, 1,19) |
0,82 (0,69, 0,99) |
| Claritromicina | 500 q12h, 7 dias | 800 três vezes ao dia, 7 dias | 10 | 1.08 (0,85, 1,38) |
1.19 (1,00, 1,42) |
1,57 (1,16, 2,12) |
| Delavirdine | 400 três vezes ao dia | 400 três vezes ao dia, 7 dias | 28 | 0,64 * (0,48, 0,86) |
Nenhuma mudança significativa * | 2.18 (1,16, 4,12) |
| Delavirdine | 400 três vezes ao dia | 600 três vezes ao dia, 7 dias | 28 | Nenhuma mudança significativa | 1,53 (1,07, 2,20) |
3,98 * (2,04, 7,78) |
| Efavirenz † | 600 uma vez ao dia, 10 dias | 1000 três vezes ao dia, 10 dias | 20 | |||
| Após a dose da manhã | Nenhuma mudança significativa * | 0,67 * (0,61, 0,74) |
0,61 * (0,49, 0,76) |
|||
| Após a dose da tarde | Nenhuma mudança significativa * | 0,63 * (0,54, 0,74) |
0,48 * (0,43, 0,53) |
|||
| Após a dose da noite | 0,71 * (0,57, 0,89) |
0,54 * (0,46, 0,63) |
0,43 * (0,37, 0,50) |
|||
| Fluconazol † | 400 uma vez ao dia, 8 dias | 1000 três vezes ao dia, 7 dias | 11 | 0,87 (0,72, 1,05) |
0,76 (0,59, 0,98) |
0,90 (0,72, 1,12) |
| Suco de toranja | 8 oz. | 400 dose única | 10 | 0,65 (0,53, 0,79) |
0,73 (0,60, 0,87) |
0,90 (0,71, 1,15) |
| Isoniazid | 300 uma vez ao dia pela manhã, 8 dias | 800 três vezes ao dia, 7 dias | 11 | 0,95 (0,88, 1,03) |
0,99 (0,87, 1,13) |
0,89 (0,75, 1,06) |
| Itraconazol | 200 duas vezes ao dia, 7 dias | 600 três vezes ao dia, 7 dias | 12 | 0,78 (0,69, 0,88) |
0,99 * (0,91, 1,06) |
1,49 (1,28, 1,74) |
| Cetoconazol | 400 uma vez ao dia, 7 dias | 600 três vezes ao dia, 7 dias | 12 | 0,69 * (0,61, 0,78) |
0,80 * (0,74, 0,87) |
1,29 (1,11, 1,51) |
| 400 uma vez ao dia, 7 dias | 400 três vezes ao dia, 7 dias | 12 | 0,42 * (0,37, 0,47) |
0,44 * (0,41, 0,48) |
0,73 * (0,62, 0,85) |
|
| Metadona | 20-60 uma vez ao dia pela manhã, 8 dias | 800 três vezes ao dia, 8 dias | 10 | Veja o texto abaixo para discussão sobre interação. | ||
| Quinidina | 200 dose única | 400 dose única | 10 | 0,96 (0,79, 1,18) |
1.07 (0,89, 1,28) |
0,93 (0,73, 1,19) |
| Rifabutin | 150 uma vez ao dia pela manhã, 10 dias | 800 três vezes ao dia, 10 dias | 14 | 0,80 (0,72, 0,89) |
0,68 (0,60, 0,76) |
0,60 (0,51, 0,72) |
| Rifabutin | 300 uma vez ao dia pela manhã, 10 dias | 800 três vezes ao dia, 10 dias | 10 | 0,75 (0,61, 0,91) |
0,66 (0,56, 0,77) |
0,61 (0,50, 0,75) |
| Rifampin | 600 uma vez ao dia pela manhã, 8 dias | 800 três vezes ao dia, 7 dias | 12 | 0,13 (0,08, 0,22) |
0,08 (0,06, 0,11) |
Não feito |
| Ritonavir | 100 duas vezes ao dia, 14 dias | 800 duas vezes ao dia, 14 dias | 10, 16 ‡ | Veja o texto abaixo para discussão sobre interação. | ||
| Ritonavir | 200 duas vezes ao dia, 14 dias | 800 duas vezes ao dia, 14 dias | 9, 16 ‡ | Veja o texto abaixo para discussão sobre interação. | ||
| Sildenafil | 25 dose única | 800 três vezes ao dia | 6 | Veja o texto abaixo para discussão sobre interação. | ||
| St. Erva de John (Hypericum perforatum, padronizada para 0,3% de hipericina) | 300 três vezes ao dia com as refeições, 14 dias | 800 três vezes ao dia | 8 | Não disponível | 0,46 (0,34, 0,58) § |
0,19 (0,06, 0,33) § |
| Stavudine (d4T) † |
40 duas vezes ao dia, 7 dias | 800 três vezes ao dia, 7 dias | 11 | 0,95 (0,80, 1,11) |
0,95 (0,80, 1,12) |
1.13 (0,83, 1,53) |
| Trimetoprim / Sulfametoxazol | 800 Trimethoprim / 160 Sulfametoxazol q12h, 7 dias | 400 quatro vezes ao dia, 7 dias | 12 | 1.12 (0,87, 1,46) |
0,98 (0,81, 1,18) |
0,83 (0,72, 0,95) |
| Zidovudina † | 200 três vezes ao dia, 7 dias | 1000 três vezes ao dia, 7 dias | 12 | 1.06 (0,91, 1,25) |
1.05 (0,86, 1,28) |
1.02 (0,77, 1,35) |
| Zidovudina / lamivudina (3TC) † |
200/150 três vezes ao dia, 7 dias | 800 três vezes ao dia, 7 dias | 6, 9¶ | 1.05 (0,83, 1,33) |
1.04 (0,67, 1,61) |
0,98 (0,56, 1,73) |
| Todos os estudos de interação realizados em saúde,
Indivíduos adultos HIV negativos, salvo indicação em contrário. * Em relação ao indinavir 800 mg três vezes ao dia sozinho. † Estudo realizado em indivíduos HIV positivos. ‡ Comparação com dados históricos de 16 indivíduos que recebem indinavir isoladamente. § IC95% . Design de grupo paralelo; n para indinavir + medicamento co-administrado, n para indinavir sozinho. |
Tabela 3: Interações medicamentosas: parâmetros farmacocinéticos
para o medicamento co-administrado na presença de indinavir (ver PRECAUTIONS, tabela 9 para
Alterações recomendadas em dose ou regime)
| Medicamento co-administrado | Dose do medicamento co-administrado (mg) | Dose de CRIXIVAN (mg) | n | Proporção (com / sem CRIXIVAN) de parâmetros farmacocinéticos de medicamentos co-administrados (IC 90%); Sem efeito = 1,00 | ||
| Cmax | AUC | Cmin | ||||
| Claritromicina | 500 duas vezes ao dia, 7 dias | 800 três vezes ao dia, 7 dias | 12 | 1.19 (1,02, 1,39) |
1,47 (1,30, 1,65) |
1,97 (1,58, 2,46) n = 11 |
| Efavirenz | 200 uma vez ao dia, 14 dias | 800 três vezes ao dia, 14 dias | 20 | Nenhuma mudança significativa | Nenhuma mudança significativa | -- |
| Etinilestradiol (ORTHO-NOVUM 1/35) * | 35 mcg, 8 dias | 800 três vezes ao dia, 8 dias | 18 | 1.02 (0,96, 1,09) |
1.22 (1,15, 1,30) |
1,37 (1,24, 1,51) |
| Isoniazid | 300 uma vez ao dia pela manhã, 8 dias | 800 três vezes ao dia, 8 dias | 11 | 1.34 (1,12, 1,60) |
1.12 (1,03, 1,22) |
1,00 (0,92, 1,08) |
| Metadona † | 20-60 uma vez ao dia pela manhã, 8 dias | 800 três vezes ao dia, 8 dias | 12 | 0,93 (0,84, 1,03) |
0,96 (0,86, 1,06) |
1.06 (0,94, 1,19) |
| Noretindrona (ORTHO-NOVUM 1/35) * |
1 mcg, 8 dias | 800 três vezes ao dia, 8 dias | 18 | 1.05 (0,95, 1,16) |
1.26 (1,20, 1,31) |
1.44 (1,32, 1,57) |
| Rifabutin 150 mg uma vez ao dia pela manhã, 11 dias + indinavir em comparação com 300 mg uma vez ao dia pela manhã, 11 dias sozinho | 150 uma vez ao dia pela manhã, 10 dias | 800 três vezes ao dia, 10 dias | 14 | 1,29 (1,05, 1,59) |
1,54 (1,33, 1,79) |
1,99 (1,71, 2,31) n = 13 |
| 300 uma vez ao dia pela manhã, 10 dias | 800 três vezes ao dia, 10 dias | 10) | 2.34 (1,64, 3,35) |
2.73 (1,99, 3,77) |
3.44 (2,65, 4,46) n = 9 |
|
| Ritonavir | 100 duas vezes ao dia, 14 dias | 800 duas vezes ao dia, 14 dias | 10, 4 ‡ | 1.61 (1,13, 2,29) |
1.72 (1,20, 2,48) |
1.62 (0,93, 2,85) |
| 200 duas vezes ao dia, 14 dias | 800 duas vezes ao dia, 14 dias | 9, 5 ‡ | 1.19 (0,85, 1,66) |
1,96 (1,39, 2,76) |
4.71 (2,66, 8,33) n = 9, 4 |
|
| Saquinavir | ||||||
| Formulação de gel duro | 600 dose única | 800 três vezes ao dia, 2 dias | 6 | 4.7 (2,7, 8,1) |
6.0 (4.0, 9.1) |
2.9 § (1,7, 4,7) |
| Formulação em gel macio Formulação em gel macio | 800 dose única 1200 dose única | 800 três vezes ao dia, 2 dias 800 três vezes ao dia, 2 dias | 6 | 6.5 (4,7, 9,1) |
7.2 (4.3, 11.9) |
5,5 § (2.2, 14.1) |
| 6 | 4.0 (2,7, 5,9) |
4.6 (3.2, 6.7) |
5,5 § (3,7, 8,3) |
|||
| Sildenafil | 25 dose única | 800 três vezes ao dia | 6 | Veja o texto abaixo para discussão sobre interação. | ||
| Stavudine¶ | 40 duas vezes ao dia, 7 dias | 800 três vezes ao dia, 7 dias | 13 | 0,86 (0,73, 1,03) |
1.21 (1,09, 1,33) |
Não feito |
| Teofilina | 250 dose única (nos dias 1 e 7) | 800 três vezes ao dia, 6 dias (Dias 2 a 7) |
12, 4 ‡ | 0,88 (0,76, 1,03) |
1.14 (1,04, 1,24) |
1.13 (0,86, 1,49) n = 7, 3 |
| Trimetoprim / Sulfametoxazol | ||||||
| Trimethoprim | 800 Trimethoprim / 160 Sulfametoxazol q12h, 7 dias | 400 q6h, 7 dias | 12 | 1.18 (1,05, 1,32) |
1.18 (1,05, 1,33) |
1.18 (1,00, 1,39) |
| Trimetoprim / Sulfametoxazol | ||||||
| Sulfametoxazol | 800 Trimethoprim / 160 Sulfametoxazol q12h, 7 dias | 400 q6h, 7 dias | 12 | 1.01 (0,95, 1,08) |
1.05 (1,01, 1,09) |
1.05 (0,97, 1,14) |
| Vardenafil | 10 dose única | 800 três vezes ao dia | 18 | Veja o texto abaixo para discussão sobre interação. | ||
| Zidovudina¶ | 200 três vezes ao dia, 7 dias | 1000 três vezes ao dia, 7 dias | 12 | 0,89 (0,73, 1,09) |
1.17 (1,07, 1,29) |
1.51 (0,71, 3,20) n = 4 |
| Zidovudina / lamivudina¶ | ||||||
| Zidovudina | 200/150 três vezes ao dia, 7 dias | 800 três vezes ao dia, 7 dias | 6, 7 ‡ | 1.23 (0,74, 2,03) |
1,39 (1,02, 1,89) |
1.08 (0,77, 1,50) n = 5, 5 |
| Zidovudina / lamivudina¶ | ||||||
| Lamivudina | 200/150 três vezes ao dia, 7 dias | 800 três vezes ao dia, 7 dias | 6, 7 ‡ | 0,73 (0,52, 1,02) |
0,91 (0,66, 1,26) |
0,88 (0,59, 1,33) |
| Todos os estudos de interação realizados em saúde,
Indivíduos adultos HIV negativos, salvo indicação em contrário. * Marca registrada da Ortho Pharmaceutical Corporation. † Estudo realizado em indivíduos sobre manutenção com metadona. ‡ Projeto paralelo do grupo; n para medicamento co-administrado + indinavir, n para medicamento administrado isoladamente. § C6hr ¶ Estudo realizado em indivíduos HIV positivos. |
Delavirdine
A delavirdina inibe o metabolismo do indinavir essa administração concomitante de 400 mg ou 600 mg de indinavir três vezes ao dia com A delavirdina de 400 mg altera três vezes ao dia a AUC, a Cmax e a Cmin do indinavir (ver quadro 2). O indinavir não teve efeito na farmacocinética da delavirdina (ver DOSAGEM E ADMINISTRAÇÃO, Terapia concomitante, Delavirdine), com base em uma comparação com dados farmacocinéticos históricos da delavirdina.
Metadona
Administração de indinavir (800 mg a cada 8 horas) com metadona (20 mg a 60 mg por dia) por uma semana em indivíduos com metadona a manutenção não resultou em alteração na AUC da metadona. Com base em uma comparação com dados históricos, houve pouca ou nenhuma alteração na AUC do indinavir
Ritonavir
Comparado aos dados históricos em pacientes que receberam indinavir 800 mg a cada 8 horas sozinho, administração concomitante duas vezes ao dia resultaram voluntários de indinavir 800 mg e ritonavir com alimentos por duas semanas num aumento de 2,7 vezes da AUC24h do indinavir, um aumento de 1,6 vezes na Cmax do indinavir e um aumento de 11 vezes na indinavir Cmin para uma dose de 100 mg de ritonavir e a Aumento de 3,6 vezes da AUC24h do indinavir, um aumento de 1,8 vezes na Cmax do indinavir e um aumento de 24 vezes na indinavir Cmin para uma dose de 200 mg de ritonavir. No mesmo estudo, administração concomitante duas vezes ao dia de indinavir (800 mg) e ritonavir (100 ou 200 mg) resultaram em aumentos de AUC24h do ritonavir versus as mesmas doses de ritonavir sozinho (ver quadro 3).
Sildenafil
Os resultados de um estudo publicado em homens infectados pelo HIV (n = 6) indicaram que a administração concomitante de indinavir (800 mg a cada 8 horas cronicamente) com uma dose única de 25 mg de sildenafil resultou em um aumento de 11% na AUC0-8hr média do indinavir e um aumento de 48% no pico médio do indinavir concentração (Cmax) em comparação com 800 mg a cada 8 horas isoladamente. Sildenafil médio A AUC aumentou 340% após a administração concomitante de sildenafil e indinavir comparado com dados históricos após a administração de sildenafil sozinho (ver CONTRA-INDICAÇÕES, AVISO, INTERAÇÕES DE DROGAS e PRECAUÇÕES: INTERAÇÕES DE DROGAS).
Vardenafil
Indinavir (800 mg a cada 8 horas) co-administrado com a uma dose única de 10 mg de vardenafil resultou em um aumento de 16 vezes no vardenafil AUC, um aumento de 7 vezes na Cmax do vardenafil e um aumento de 2 vezes no vardenafil meia-vida (ver AVISO, INTERAÇÕES DE DROGAS e PRECAUÇÕES: INTERAÇÕES DE DROGAS).
Categoria de gravidez C: Toxicidade para o desenvolvimento foram realizados estudos em coelhos (em doses de até 240 mg / kg / dia), cães (em doses de até 80 mg / kg / dia) e ratos (em doses de até 640 mg / kg / dia). O mais alto doses nesses estudos produziram exposições sistêmicas nessas espécies comparáveis ou ligeiramente superior à exposição humana. Nenhum externo relacionado ao tratamento alterações viscerais ou esqueléticas foram observadas em coelhos ou cães. Não alterações externas ou viscerais relacionadas ao tratamento foram observadas em ratos. Aumentos relacionados ao tratamento sobre controles na incidência de costelas supranumerárias (em exposições iguais ou inferiores às dos seres humanos) e de costelas cervicais (em exposições comparáveis ou ligeiramente superiores aos dos seres humanos) foram observados em ratos. No todas as três espécies, sem efeitos relacionados ao tratamento na sobrevida embrionária / fetal ou pesos fetais foram observados.
Nos coelhos, na dose materna de 240 mg / kg / dia, nenhum medicamento foi detectado no plasma fetal 1 hora após a administração. Níveis fetais de medicamentos no plasma 2 horas após a administração foram aproximadamente 3% dos níveis maternos de medicamentos plasmáticos. No cães, na dose materna de 80 mg / kg / dia, os níveis plasmáticos fetais foram aproximadamente 50% dos níveis de drogas plasmáticas maternas 1 e 2 horas depois dosagem. Em ratos, em doses maternas de 40 e 640 mg / kg / dia, medicamento plasmático fetal os níveis foram de aproximadamente 10 a 15% e 10 a 20% do medicamento materno para plasma níveis 1 e 2 horas após a administração, respectivamente.
O indinavir foi administrado a macacos Rhesus durante o terceiro trimestre de gravidez (em doses de até 160 mg / kg duas vezes ao dia) e para macacos Rhesus neonatais (em doses de até 160 mg / kg duas vezes ao dia). Quando administrado para os neonatos, o indinavir causou uma exacerbação da hiperbilirrubinemia fisiológica transitória visto nesta espécie após o nascimento; os valores séricos de bilirrubina foram aproximadamente quatro vezes acima dos controles a 160 mg / kg duas vezes ao dia. Uma exacerbação semelhante fez não ocorrer em neonatos após exposição no útero ao indinavir durante o terceiro trimestre de gravidez. Nos macacos Rhesus, os níveis fetais de drogas plasmáticas eram aproximadamente 1 a 2% dos níveis de drogas plasmáticas maternas aproximadamente 1 hora depois dosagem materna a 40, 80 ou 160 mg / kg duas vezes ao dia.
Ocorreu hiperbilirrubinemia durante o tratamento com CRIXIVAN (Vejo PRECAUÇÕES e REAÇÕES ADVERSAS). Não se sabe se CRIXIVAN administrado à mãe no período perinatal irá exacerbar hiperbilirrubinemia fisiológica em neonatos.
Não há estudos adequados e bem controlados pacientes grávidas. CRIXIVAN deve ser utilizado durante a gravidez apenas se o o benefício potencial justifica o risco potencial para o feto.
Uma dose de CRIXIVAN de 800 mg a cada 8 horas (com zidovudina 200 mg a cada 8 horas e lamivudina 150 mg duas vezes ao dia) foi estudado em 16 infectados pelo HIV pacientes grávidas com 14 a 28 semanas de gestação na inscrição (estudo PACTG 358). Dadas as exposições antepartum substancialmente mais baixas observadas e as limitadas dados nesta população de pacientes, o uso de indinavir não é recomendado Pacientes grávidas infectadas pelo HIV (ver FARMACOLOGIA CLÍNICA, Grávida Pacientes).
Registro de Gravidez Anti-retroviral
Monitorar os resultados materno-fetais de pacientes grávidas exposto ao CRIXIVAN, foi estabelecido um Registro de Gravidez Anti-retroviral. Os médicos são incentivados a registrar pacientes ligando para 1-800-258-4263.
Ensaios clínicos em adultos
Nefrolitíase / urolitíase, incluindo dor no flanco ou sem hematúria (incluindo hematúria microscópica), foi relatado em aproximadamente 12,4% (301/2429; intervalo entre ensaios individuais: 4,7% a 34,4%) de doentes a receber CRIXIVAN na dose recomendada em ensaios clínicos com um acompanhamento médio de 47 semanas (intervalo: 1 dia a 242 semanas; 2238 pacientes-ano acompanhamento). A frequência cumulativa de eventos de nefrolitíase aumenta com duração da exposição ao CRIXIVAN; no entanto, o risco ao longo do tempo permanece relativamente constante. Dos pacientes tratados com CRIXIVAN que se desenvolveram nefrolitíase / urolitíase em ensaios clínicos durante a fase duplo-cega, Foi relatado que 2,8% (7/246) desenvolvem hidronefrose e 4,5% (11/246) foi submetido a colocação de stent. Após o episódio agudo, 4,9% (12/246) de pacientes interromperam a terapia. (Vejo AVISO e DOSAGEM E ADMINISTRAÇÃO, Nefrolitíase / Urolitíase.)
Hiperbilirrubinemia assintomática (bilirrubina total ≥ 2,5 mg / dL), relatado predominantemente como bilirrubina indireta elevada, possui ocorreu em aproximadamente 14% dos pacientes tratados com CRIXIVAN. Em <1% isso foi associado a elevações em ALT ou AST
Hiperbilirrubinemia e nefrolitíase / urolitíase ocorreu com mais frequência em doses superiores a 2,4 g / dia em comparação com doses ≤ 2,4 g / dia.
Experiências adversas clínicas relatadas em ≥ 2% de doentes tratados apenas com CRIXIVAN, CRIXIVAN em associação com zidovudina ou zidovudina mais lamivudina, zidovudina isoladamente ou zidovudina mais lamivudina são apresentados na Tabela 10.
Tabela 10: Experiências clínicas adversas relatadas em ≥ 2%
de pacientes
| Experiência Adversa | Estudo 028 Considerado relacionado a medicamentos e de intensidade moderada ou grave | Estudo ACTG 320 de Relações com Drogas Desconhecidas e de Intensidade Grave ou com Ameaça de Vida | |||
| Porcentagem CRIXIVAN (n = 332) |
CRIXIVAN mais porcentagem de zidovudina (n = 332) |
Porcentagem de zidovudina (n = 332) |
CRIXIVAN mais Zidovudina mais porcentagem de lamivudina (n = 571) |
Zidovudina mais porcentagem de lamivudina (n = 575) |
|
| Corpo como um todo | |||||
| Dor abdominal | 16,6 | 16,0 | 12,0 | 1.9 | 0,7 |
| Astenia / fadiga | 2.1 | 4.2 | 3.6 | 2.4 | 4.5 |
| Febre | 1.5 | 1.5 | 2.1 | 3.8 | 3.0 |
| Mal-estar | 2.1 | 2.7 | 1.8 | 0 | 0 |
| Sistema Digestivo | |||||
| Náusea | 11,7 | 31,9 | 19,6 | 2.8 | 1.4 |
| Diarréia | 3.3 | 3.0 | 2.4 | 0,9 | 1.2 |
| Vômitos | 8.4 | 17,8 | 9.0 | 1.4 | 1.4 |
| Regurgitação ácida | 2.7 | 5.4 | 1.8 | 0.4 | 0 |
| Anorexia | 2.7 | 5.4 | 3.0 | 0,5 | 0.2 |
| Aumento de apetite | 2.1 | 1.5 | 1.2 | 0 | 0 |
| Dispepsia | 1.5 | 2.7 | 0,9 | 0 | 0 |
| Icterícia | 1.5 | 2.1 | 0.3 | 0 | 0 |
| Sistema Hêmico e Linfático | |||||
| Anemia | 0.6 | 1.2 | 2.1 | 2.4 | 3.5 |
| Sistema músculo-esquelético | |||||
| Dor nas costas | 8.4 | 4.5 | 1.5 | 0,9 | 0,7 |
| Sistema Nervoso / Psiquiátrico | |||||
| Dor de cabeça | 5.4 | 9.6 | 6.0 | 2.4 | 2.8 |
| Tontura | 3.0 | 3.9 | 0,9 | 0,5 | 0,7 |
| Sonolência | 2.4 | 3.3 | 3.3 | 0 | 0 |
| Apêndices de pele e pele | |||||
| Prurido | 4.2 | 2.4 | 1.8 | 0,5 | 0 |
| Erupção cutânea | 1.2 | 0.6 | 2.4 | 1.1 | 0,5 |
| Sistema Respiratório | |||||
| Tosse | 1.5 | 0.3 | 0.6 | 1.6 | 1.0 |
| Dificuldade em respirar / dispnéia / falta de ar | 0 | 0.6 | 0.3 | 1.8 | 1.0 |
| Sistema Urogenital | |||||
| Nefrolitíase / urolitíase | 8.7 | 7.8 | 2.1 | 2.6 | 0.3 |
| Disúria | 1.5 | 2.4 | 0.3 | 0.4 | 0.2 |
| Sentidos especiais | |||||
| Proversão de sabor | 2.7 | 8.4 | 1.2 | 0.2 | 0 |
| * Incluindo cólica renal e dor no flanco com e sem hematúria | |||||
Nas fases I e II controladas ensaios, os seguintes eventos adversos foram relatados significativamente mais freqüentemente por aqueles randomizados para os braços que contêm CRIXIVAN do que por aqueles randomizado para análogos de nucleosídeos: erupção cutânea, infecção respiratória superior, seca pele, faringite, perversão do paladar.
Laboratório selecionado anormalidades de intensidade grave ou com risco de vida relatadas em pacientes tratados apenas com CRIXIVAN, CRIXIVAN em combinação com zidovudina ou são zidovudina mais lamivudina, zidovudina isoladamente ou zidovudina mais lamivudina apresentado na Tabela 11.
Tabela 11: Anormalidades laboratoriais selecionadas de gravidade
ou Intensidade com risco de vida relatada nos estudos 028 e ACTG 320
| Estudo 028 Considerado relacionado a medicamentos e de intensidade moderada ou grave | Estudo ACTG 320 de Relações com Drogas Desconhecidas e de Intensidade Grave ou com Ameaça de Vida | ||||
| Porcentagem CRIXIVAN (n = 329) |
CRIXIVAN mais porcentagem de zidovudina (n = 320) |
Porcentagem de zidovudina (n = 330) |
CRIXIVAN mais Zidovudina mais porcentagem de lamivudina (n = 571) |
Zidovudina mais porcentagem de lamivudina (n = 575) |
|
| Hematologia | |||||
| Diminuição da hemoglobina <7,0 g / dL | 0.6 | 0,9 | 3.3 | 2.4 | 3.5 |
| Diminuição da contagem de plaquetas <50 THS / mm³ | 0,9 | 0,9 | 1.8 | 0.2 | 0,9 |
| Neutrófilos diminuídos <0,75 THS / mm³ | 2.4 | 2.2 | 6.7 | 5.1 | 14,6 |
| Química do sangue | |||||
| ALT aumentado> 500% LSN * | 4.9 | 4.1 | 3.0 | 2.6 | 2.6 |
| Aumento de AST> 500% LSN | 3.7 | 2.8 | 2.7 | 3.3 | 2.8 |
| Bilirrubina sérica total> 250% LSN | 11,9 | 9.7 | 0.6 | 6.1 | 1.4 |
| Aumento da amilase sérica> 200% LSN | 2.1 | 1.9 | 1.8 | 0,9 | 0.3 |
| Aumento da glicose> 250 mg / dL | 0,9 | 0,9 | 0.6 | 1.6 | 1.9 |
| Creatinina aumentada> 300% LSN | 0 | 0 | 0.6 | 0.2 | 0 |
| * Limite superior do normal alcance. | |||||
Experiência pós-comercialização
Corpo como um todo : redistribuição / acumulação de gordura corporal (ver PRECAUÇÕES, Redistribuição de gordura).
Sistema Cardiovascular: distúrbios cardiovasculares incluindo infarto do miocárdio e angina de peito; distúrbio cerebrovascular.
Sistema Digestivo: função hepática anormalidades; hepatite, incluindo relatos de insuficiência hepática (ver AVISO); pancreatite; icterícia; distensão abdominal; dispepsia.
Hematológico: aumentado espontâneo hemorragia em doentes com hemofilia (ver PRECAUÇÕES); hemolítico agudo anemia (ver AVISO).
Endócrino / metabólico : novo diabetes mellitus, exacerbação do diabetes mellitus preexistente, hiperglicemia (ver AVISO).
Hipersensibilidade: reações anafilactóides; urticária; vasculite.
Sistema músculo-esquelético : artralgia, periartrite.
Sistema Nervoso / Psiquiatria : parestesia oral; depressão.
Apêndices de pele e pele : erupção cutânea incluindo eritema síndrome de multiforme e Stevens-Johnson; hiperpigmentação; alopecia; encravado unhas dos pés e / ou paroníquia; prurido.
Sistema Urogenital : nefrolitíase / urolitíase, em alguns casos, resultando em insuficiência renal ou insuficiência renal aguda pielonefrite com ou sem bacteremia (ver AVISO); intersticial nefrite às vezes com depósitos de cristal de indinavir; em alguns pacientes, o a nefrite intersticial não se resolveu após a descontinuação do CRIXIVAN ; insuficiência renal; insuficiência renal; leucocitúria (ver PRECAUÇÕES), cristalúria; disúria.
Anormalidades laboratoriais
Triglicerídeos séricos aumentados; aumento do colesterol sérico.
Houve mais de 60 relatos de agudos ou crônicos superdosagem humana (até 23 vezes a dose diária total recomendada de 2400 mg) com CRIXIVAN. Os sintomas mais comumente relatados foram renais (por exemplo,., nefrolitíase / urolitíase, dor no flanco, hematúria) e gastrointestinal (por exemplo., náusea, vômito, diarréia).
Não se sabe se o CRIXIVAN é dializável por peritoneal ou hemodiálise.
Absorção
O indinavir foi rapidamente absorvido no estado de jejum com a tempo até o pico da concentração plasmática (Tmax) de 0,8 ± 0,3 horas (média ± S.D.) (n = 11). Um aumento superior à proporcional à dose no plasma de indinavir foram observadas concentrações no intervalo de doses de 200-1000 mg. Em uma dosagem regime de 800 mg a cada 8 horas, área de estado estacionário sob o plasma a curva do tempo de concentração (AUC) foi de 30.691 ± 11.407 nM ™ ¬ ¢ hora (n = 16), pico do plasma a concentração (Cmax) foi de 12.617 ± 4037 nM (n = 16) e a concentração plasmática oito horas após a dose (seca) foi de 251 ± 178 nM (n = 16).
Efeito dos alimentos na absorção oral
Administração de indinavir com uma refeição rica em calorias gordura e proteína (784 kcal, 48,6 g de gordura, 31,3 g de proteína) resultaram em 77% ± 8% redução na AUC e uma redução de 84% ± 7% na Cmax (n = 10). Administração com refeições mais leves (por exemplo,., uma refeição de torradas secas com geléia, suco de maçã e café com leite desnatado e açúcar ou uma refeição de flocos de milho, leite desnatado e açúcar) resultou em pouca ou nenhuma alteração na concentração de AUC, Cmax ou vale.
Distribuição
O indinavir estava aproximadamente 60% ligado ao plasma humano proteínas em uma faixa de concentração de 81 nM a 16.300 nM
Metabolismo
Após uma dose de 400 mg de 14C-indinavir, 83 ± 1% (n = 4) e 19 ± 3% (n = 6) da radioatividade total foram recuperados nas fezes e na urina respectivamente; a radioatividade devido ao medicamento original nas fezes e na urina foi de 19,1% e 9,4%, respectivamente. Sete metabólitos foram identificados, um conjugado glucuronídeo e seis metabólitos oxidativos. In vitro estudos indicam que o citocromo P-450 3A4 (CYP3A4) é a principal enzima responsável pela formação do oxidativo metabolitos.
Eliminação
Menos de 20% do indinavir é excretado inalterado no urina. A excreção urinária média do medicamento inalterado foi de 10,4 ± 4,9% (n = 10) e 12,0 ± 4,9% (n = 10) após uma dose única de 700 mg e 1000 mg, respectivamente. O indinavir foi rapidamente eliminado com uma meia-vida de 1,8 ± 0,4 horas (n = 10). Acúmulo significativo não foi observado após doses múltiplas a 800 mg a cada 8 horas.
Disponível em países
 Australia
Australia Austria
Austria Belgium
Belgium Brazil
Brazil Bulgaria
Bulgaria Canada
Canada China
China Cyprus
Cyprus Czech Republic
Czech Republic Denmark
Denmark Estonia
Estonia Finland
Finland France
France Germany
Germany Greece
Greece Hong Kong
Hong Kong Hungary
Hungary Iceland
Iceland Ireland
Ireland Italy
Italy Japan
Japan Latvia
Latvia Lebanon
Lebanon Liechtenstein
Liechtenstein Lithuania
Lithuania Luxembourg
Luxembourg Malta
Malta Netherlands
Netherlands New Zealand
New Zealand Norway
Norway Philippines
Philippines Poland
Poland Portugal
Portugal Romania
Romania Russia
Russia Serbia
Serbia Slovakia
Slovakia Slovenia
Slovenia South Africa
South Africa Spain
Spain Sweden
Sweden Switzerland
Switzerland Taiwan
Taiwan Turkey
Turkey United Kingdom
United Kingdom USA
USA Venezuela
Venezuela