


Composition:
Application:
Utilisé dans le traitement:
Examiné médicalement par Oliinyk Elizabeth Ivanovna, Pharmacie Dernière mise à jour le 10.04.2022

Attention! Information sur la page est réservée aux professionnels de la santé! Les informations sont collectées dans des sources ouvertes et peuvent contenir des erreurs significatives! Soyez prudent et revérifiez toutes les informations de cette page!
Formes posologiques et forces
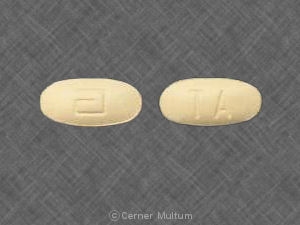
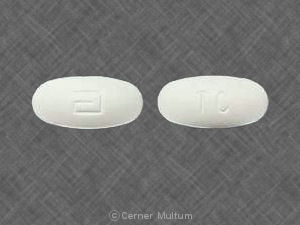
- Comprimés jaunes à 54 mg, imprimés avec le logo «a» et les lettres d'identification de code «TA».
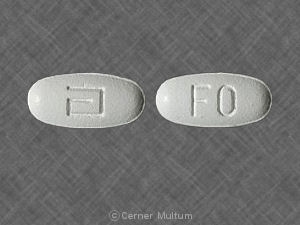
- Comprimés blancs à 160 mg, imprimés avec le logo «a» et les lettres d'identification de code «TC».
Stockage et manutention
TRICOR® (comprimés de fénofibrate) est disponible en deux points forts:
54 mg comprimés jaunes, imprimés avec le logo «a» et les lettres d'identification de code «TA», disponibles en flacons de 90 (NDC 0074-4009-90).
160 mg comprimés blancs, imprimés avec le logo «a» et les lettres d'identification de code «TC», disponibles en flacons de 90 (NDC 0074-4013-90).
Stockage
Conserver à 25 ° C (77 ° F); excursions autorisées à 15-30 ° C (59-86 ° F).
Tenir hors de portée des enfants. Protéger de l'humidité.
Fabriqué par: Laboratoires Fournier, S.A., 21300 Chenôve, France. Révisé: janvier 2017.
Hypercholestérolémie primaire ou dyslipidémie mixte
TRICOR est indiqué comme traitement d'appoint au régime alimentaire pour réduire le cholestérol élevé des lipoprotéines de basse densité (LDL-C), le cholestérol total (Total-C), les triglycérides et l'apolipoprotéine B (Apo B), et pour augmenter le cholestérol lipoprotéique de haute densité (HDL-C) chez les patients adultes atteints d'hypercholestérol primaire ou de dyslipidémie.
Hypertriglycéridémie sévère
TRICOR est également indiqué comme traitement d'appoint au régime alimentaire pour le traitement des patients adultes atteints d'hypertriglycéridémie sévère. L'amélioration du contrôle glycémique chez les patients diabétiques présentant une chylomicronémie à jeun évitera généralement le besoin d'intervention pharmacologique.
Niveaux sensiblement élevés de triglycérides sériques (par ex. > 2 000 mg / dL) peut augmenter le risque de développer une pancréatite. L'effet du traitement par fénofibrate sur la réduction de ce risque n'a pas été suffisamment étudié.
Limitations d'utilisation importantes
Il n'a pas été démontré que le fénofibrate à une dose équivalente à 160 mg de TRICOR réduit la morbidité et la mortalité des maladies coronariennes dans un grand essai contrôlé randomisé de patients atteints de diabète sucré de type 2.
Considérations générales
Les patients doivent suivre un régime approprié pour réduire les lipides avant de recevoir TRICOR et doivent poursuivre ce régime pendant le traitement par TRICOR. Les comprimés TRICOR doivent être administrés avec les repas, optimisant ainsi la biodisponibilité du médicament.
Le traitement initial de la dyslipidémie est une thérapie alimentaire spécifique au type d'anomalie des lipoprotéines. L'excès de poids corporel et l'apport alcoolique excessif peuvent être des facteurs importants de l'hypertriglycéridémie et doivent être traités avant tout traitement médicamenteux. L'exercice physique peut être une mesure accessoire importante. Les maladies contribuant à l'hyperlipidémie, telles que l'hypothyroïdie ou le diabète sucré, doivent être recherchées et traitées de manière adéquate. La thérapie aux œstrogènes, les diurétiques thiazidiques et les bêta-bloquants sont parfois associés à des augmentations massives des triglycérides plasmatiques, en particulier chez les sujets atteints d'hypertriglycéridémie familiale. Dans de tels cas, l'arrêt de l'agent étiologique spécifique peut éviter la nécessité d'un traitement médicamenteux spécifique de l'hypertriglycéridémie.
Les taux de lipides doivent être surveillés périodiquement et il convient de prendre en compte la réduction de la posologie de TRICOR si les niveaux de lipides tombent nettement en dessous de la plage cible.
Le traitement doit être retiré chez les patients qui n'ont pas de réponse adéquate après deux mois de traitement avec la dose maximale recommandée de 160 mg une fois par jour.
Hypercholestérolémie primaire ou dyslipidémie mixte
La dose initiale de TRICOR est de 160 mg une fois par jour.
Hypertriglycéridémie sévère
La dose initiale est de 54 à 160 mg par jour. La posologie doit être individualisée en fonction de la réponse du patient et doit être ajustée si nécessaire après des déterminations répétées des lipides à des intervalles de 4 à 8 semaines. La dose maximale est de 160 mg une fois par jour.
Fonction rénale altérée
Le traitement par TRICOR doit être initié à une dose de 54 mg par jour chez les patients présentant une fonction rénale légère à modérément altérée et augmenté uniquement après évaluation des effets sur la fonction rénale et les taux de lipides à cette dose. L'utilisation de TRICOR doit être évitée chez les patients présentant une insuffisance rénale sévère.
Patients gériatriques
La sélection de la dose pour les personnes âgées doit être effectuée en fonction de la fonction rénale.
TRICOR est contre-indiqué dans :
- les patients présentant une insuffisance rénale sévère, y compris ceux recevant une dialyse.
- les patients atteints d'une maladie hépatique active, y compris ceux atteints de cirrhose biliaire primaire et anomalies inexpliquées de la fonction hépatique persistante.
- patients atteints d'une maladie préexistante de la vésicule biliaire.
- mères allaitantes
- patients présentant une hypersensibilité connue au fénofibrate ou à l'acide fénofibrique.
AVERTISSEMENTS
Inclus dans le cadre du "PRECAUTIONS" Section
PRÉCAUTIONS
Morbidité de la mortalité et des maladies coronariennes
L'effet de TRICOR sur la morbidité et la mortalité coronariennes et la mortalité non cardiovasculaire n'a pas été établi.
L'essai Action pour contrôler le risque cardiovasculaire chez le lipide diabétique (ACCORD Lipid) était une étude randomisée contrôlée contre placebo portant sur 5518 patients atteints de diabète sucré de type 2 sous statine de fond traités par fénofibrate. La durée moyenne du suivi était de 4,7 ans. La thérapie combinée Fénofibrate plus statine a montré une réduction non significative du risque relatif de 8% dans le résultat principal des événements cardiovasculaires indésirables majeurs (MACE), un composite d'infarctus du myocarde non mortel, d'un AVC non mortel et de décès par maladie cardiovasculaire (rapport de risque [HR] 0,92, IC 95% 0,79-1,08-1,08) 2. Dans une analyse de sous-groupe de genre, le rapport de risque pour MACE chez les hommes recevant une thérapie combinée par rapport à la monothérapie à la statine était de 0,82 (IC à 95% 0,69-0,99), et le rapport de risque pour MACE chez les femmes recevant une thérapie combinée par rapport à la monothérapie à la statine était de 1,38 (IC à 95% 0,98%). La signification clinique de cette découverte de sous-groupe n'est pas claire.
L'étude sur l'intervention en fénofibrate et l'abaissement des événements dans le diabète (FIELD) était une étude randomisée de 5 ans contrôlée contre placebo portant sur 9795 patients atteints de diabète sucré de type 2 traités par fénofibrate. Le fénofibrate a démontré une réduction relative non significative de 11% du résultat primaire des événements coronariens de maladies cardiaques (rapport de risque [HR] 0,89, IC 95% 0,75-1,05, p = 0,16) et une réduction significative de 11% du résultat secondaire de total événements de maladies cardiovasculaires (HR 0,89 [0,80,80,80-. Il y a eu une augmentation non significative de 11% (HR 1,11 [0,95, 1,29], p = 0,18) et 19% (HR 1,19 [0,90, 1,57], p = 0,22) de la mortalité cardiaque totale et coronaire, respectivement, avec le fénofibrate par rapport au placebo.
En raison des similitudes chimiques, pharmacologiques et cliniques entre TRICOR (comprimés de fénofibrate), clofibrate et gemfibrozil, les résultats indésirables dans 4 grandes études cliniques randomisées contrôlées contre placebo avec ces autres médicaments contre le fibrate peuvent également s'appliquer à TRICOR
Dans le Coronary Drug Project, une grande étude sur l'infarctus post-myocardique des patients traités pendant 5 ans avec du clofibrate, il n'y avait aucune différence de mortalité observée entre le groupe clofibrate et le groupe placebo. Il y avait cependant une différence dans le taux de cholélithiase et de cholécystite nécessitant une intervention chirurgicale entre les deux groupes (3,0% contre. 1,8%).
Dans une étude menée par l'Organisation mondiale de la santé (OMS), 5000 sujets sans maladie coronarienne connue ont été traités par placebo ou clofibrate pendant 5 ans et suivis pendant un an supplémentaire. Il y avait une mortalité toutes causes confondues statistiquement significative et ajustée selon l'âge dans le groupe clofibrate par rapport au groupe placebo (5,70% contre. 3,96%, p = <0,01). L'excès de mortalité est dû à une augmentation de 33% des causes non cardiovasculaires, notamment la malignité, les complications post-cholécystectomiques et la pancréatite. Cela semble confirmer le risque plus élevé de maladie de la vésicule biliaire observé chez les patients traités par le clofibrate étudiés dans le cadre du projet de médicament coronaire.
L'étude du cœur d'Helsinki était une grande étude (n = 4081) sur des hommes d'âge moyen sans antécédents de maladie coronarienne. Les sujets ont reçu un placebo ou un gemfibrozil pendant 5 ans, avec une extension ouverte de 3,5 ans par la suite. La mortalité totale était numériquement plus élevée dans le groupe de randomisation gemfibrozil mais n'a pas atteint de signification statistique (p = 0,19, intervalle de confiance à 95% pour le risque relatif G: P = 0,91-1,64). Bien que les décès par cancer aient tendance à augmenter dans le groupe gemfibrozil (p = 0,11), les cancers (à l'exclusion du carcinome basocellulaire) ont été diagnostiqués avec une fréquence égale dans les deux groupes d'étude. En raison de la taille limitée de l'étude, le risque relatif de décès d'une cause n'a pas été démontré différent de celui observé dans les données de suivi sur 9 ans de l'étude de l'Organisation mondiale de la santé (RR = 1,29).
Un élément de prévention secondaire de l'étude cardiaque d'Helsinki a recruté des hommes d'âge moyen exclus de l'étude de prévention primaire en raison d'une maladie coronarienne connue ou suspectée. Les sujets ont reçu du gemfibrozil ou un placebo pendant 5 ans. Bien que les décès cardiaques aient tendance à augmenter dans le groupe gemfibrozil, cela n'était pas statistiquement significatif (rapport de risque 2,2, intervalle de confiance à 95%: 0,94-5,05). Le taux de chirurgie de la vésicule biliaire n'était pas statistiquement significatif entre les groupes d'étude, mais il a tendance à augmenter dans le groupe gemfibrozil (1,9% contre. 0,3%, p = 0,07).
Muscle squelettique
Les fibrates augmentent le risque de myopathie et ont été associés à la rhabdomyolyse. Le risque de toxicité musculaire grave semble augmenter chez les patients âgés et chez les patients diabétiques, d'insuffisance rénale ou d'hypothyroïdie.
La myopathie doit être envisagée chez tout patient présentant des myalgies diffuses, une sensibilité ou une faiblesse musculaire et / ou des élévations marquées des taux de créatine phosphokinase (CPK).
Il faut conseiller aux patients de signaler rapidement des douleurs musculaires, une sensibilité ou une faiblesse inexpliquées, en particulier si elles sont accompagnées de malaise ou de fièvre. Les taux de CPK doivent être évalués chez les patients signalant ces symptômes, et le traitement par TRICOR doit être interrompu si des taux de CPK nettement élevés se produisent ou si une myopathie / myosite est suspectée ou diagnostiquée.
Les données des études observationnelles indiquent que le risque de rhabdomyolyse augmente lorsque les fibrates, en particulier le gemfibrozil, sont co-administrés avec un inhibiteur de l'HMG-CoA réductase (statine). La combinaison doit être évitée à moins que le bénéfice de nouvelles modifications des niveaux de lipides ne l'emporte probablement sur le risque accru de cette combinaison de médicaments.
Des cas de myopathie, y compris une rhabdomyolyse, ont été rapportés avec des fénofibrates co-administrés avec de la colchicine, et la prudence est de mise lors de la prescription de fénofibrate avec de la colchicine.
Fonction hépatique
Le fénofibrate à des doses équivalentes à 107 mg à 160 mg TRICOR par jour a été associé à des augmentations des transaminases sériques [AST (SGOT) ou ALT (SGPT)]. Dans une analyse groupée de 10 essais contrôlés contre placebo, des augmentations à> 3 fois la limite supérieure de la normale sont survenues chez 5,3% des patients prenant du fénofibrate contre 1,1% des patients traités par placebo.
Lorsque les déterminations de transaminases ont été suivies soit après l'arrêt du traitement, soit pendant la poursuite du traitement, un retour aux limites normales a généralement été observé. L'incidence des augmentations des transaminases liées au traitement par le fénofibrate semble être liée à la dose. Dans une étude de 8 semaines sur l'administration de doses, l'incidence des élévations de l'ALAT ou de l'AST à au moins trois fois la limite supérieure de la normale était de 13% chez les patients recevant des doses équivalentes à 107 mg à 160 mg TRICOR par jour et était de 0% chez ceux recevant des doses équivalentes à 54 mg ou moins TRICOR par jour, ou placebo. Une hépatite hépatocellulaire, chronique active et cholestatique associée à un traitement au fénofibrate a été rapportée après des expositions de semaines à plusieurs années. Dans de très rares cas, une cirrhose a été rapportée en association avec une hépatite active chronique.
Une surveillance périodique de base et régulière de la fonction hépatique, y compris l'ALAT sérique (SGPT) doit être effectuée pendant la durée du traitement par TRICOR, et le traitement doit être interrompu si les taux d'enzymes persistent au-dessus de trois fois la limite normale.
Créatinine sérique
Des élévations de la créatinine sérique ont été rapportées chez des patients sous fénofibrate. Ces élévations ont tendance à revenir à la ligne de base après l'arrêt du fénofibrate. La signification clinique de ces observations est inconnue. Surveiller la fonction rénale chez les patients atteints d'insuffisance rénale prenant TRICOR. Une surveillance rénale doit également être envisagée pour les patients prenant TRICOR à risque d'insuffisance rénale tels que les personnes âgées et les patients diabétiques.
Cholélithiase
Le fénofibrate, comme le clofibrate et le gemfibrozil, peut augmenter l'excrétion de cholestérol dans la bile, entraînant une cholélithiase. Si une cholélithiase est suspectée, des études sur la vésicule biliaire sont indiquées. Le traitement par TRICOR doit être interrompu si des calculs biliaires sont trouvés.
Coumarin Anticoagulants
Il faut être prudent lorsque des anticoagulants coumarins sont administrés en association avec TRICOR en raison de la potentialisation des effets anticoagulants de type coumarine dans la prolongation du temps de prothrombine / rapport international normalisé (PT / INR). Pour prévenir les complications hémorragiques, une surveillance fréquente du PT / INR et un ajustement de la dose de l'anticoagulant sont recommandés jusqu'à ce que le PT / INR se soit stabilisé.
Pancréatite
Une pancréatite a été rapportée chez des patients prenant du fénofibrate, du gemfibrozil et du clofibrate. Cette occurrence peut représenter un échec d'efficacité chez les patients atteints d'hypertriglycéridémie sévère, d'un effet médicamenteux direct ou d'un phénomène secondaire médié par la formation de pierres ou de boues des voies biliaires avec obstruction du canal biliaire commun.
Changements hématologiques
Des diminutions légères à modérées de l'hémoglobine, de l'hématocrite et des globules blancs ont été observées chez les patients après le début du traitement par le fénofibrate. Cependant, ces niveaux se stabilisent pendant l'administration à long terme. Une thrombocytopénie et une agranulocytose ont été rapportées chez des personnes traitées par le fénofibrate. Une surveillance périodique du nombre de globules rouges et blancs est recommandée pendant les 12 premiers mois d'administration de TRICOR.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité aiguës telles que le syndrome de Stevens-Johnson et une nécrolyse épidermique toxique nécessitant une hospitalisation des patients et un traitement par des stéroïdes ont été rapportées chez des personnes traitées par des fénofibrates. L'urticaire a été vue en 1.1 contre. 0% et éruption cutanée en 1,4 contre. 0,8% des patients atteints de fénofibrate et de placebo respectivement dans les essais contrôlés.
Maladie vénothromboembolique
Dans l'essai FIELD, une embolie pulmonaire (PE) et une thrombose veineuse profonde (TVP) ont été observées à des taux plus élevés dans le fénofibrate que dans le groupe traité par placebo. Sur 9 795 patients inscrits au FIELD, il y en avait 4 900 dans le groupe placebo et 4 895 dans le groupe fénofibrate. Pour le DVT, il y a eu 48 événements (1%) dans le groupe placebo et 67 (1%) dans le groupe fénofibrate (p = 0,074); et pour PE, il y a eu 32 (0,7%) événements dans le groupe placebo et 53 (1%) dans le groupe fénofibrate (p = 0,0,0222).
Dans le projet de médicament coronaire, une proportion plus élevée du groupe clofibrate a connu une embolie pulmonaire ou une thrombophlébite mortelle ou non mortelle définie ou suspectée que le groupe placebo (5,2% contre. 3,3% à cinq ans; p <0,01).
Diminutions paradoxales des niveaux de cholestérol HDL
Des rapports d'essais cliniques et post-commercialisation ont fait état de fortes diminutions des taux de cholestérol HDL (aussi faibles que 2 mg / dL) survenant chez des patients diabétiques et non diabétiques initiés au traitement par fibrate. La diminution du HDL-C se reflète dans une diminution de l'apolipoprotéine A1. Cette diminution s'est produite dans les 2 semaines à quelques années suivant le début du traitement par le fibrate. Les niveaux de HDL-C restent déprimés jusqu'à ce que le traitement par fibrate ait été retiré; la réponse au retrait du traitement par fibrate est rapide et soutenue. La signification clinique de cette diminution du HDL-C est inconnue. Il est recommandé de vérifier les niveaux de HDL-C dans les premiers mois suivant le début du traitement par fibrate. Si un niveau HDL-C gravement déprimé est détecté, le traitement par fibrate doit être retiré et le niveau HDL-C surveillé jusqu'à ce qu'il soit revenu à la ligne de base, et le traitement par fibrate ne doit pas être relancé.
Toxicologie non clinique
Cancérogenèse et mutagenèse et altération de la fertilité
Deux études de cancérogénicité alimentaire ont été menées chez le rat atteint de fénofibrate. Dans la première étude de 24 mois, les rats Wistar ont reçu une dose de fénofibrate à 10, 45 et 200 mg / kg / jour, environ 0,3, 1 et 6 fois la dose humaine maximale recommandée (MRHD), sur la base de comparaisons de la surface corporelle (mg / m2). À une dose de 200 mg / kg / jour (à 6 fois le MRHD), l'incidence des carcinomes hépatiques a été significativement augmentée chez les deux sexes. Une augmentation statistiquement significative des carcinomes pancréatiques a été observée chez les hommes à 1 et 6 fois le MRHD; une augmentation des adénomes pancréatiques et des tumeurs bénignes des cellules interstitielles testiculaires a été observée à 6 fois le MRHD chez les hommes. Dans une deuxième étude de cancérogénicité chez le rat de 24 mois dans une souche différente de rats (Sprague-Dawley) doses de 10 et 60 mg / kg / jour (0,3 et 2 fois le MRHD) a produit des augmentations significatives de l'incidence des adénomes d'acinars pancréatiques chez les deux sexes et des augmentations des tumeurs des cellules interstitielles testiculaires chez les hommes à 2 fois le MRHD
Une étude de cancérogénicité de 117 semaines a été menée chez le rat comparant trois médicaments: le fénofibrate 10 et 60 mg / kg / jour (0,3 et 2 fois le MRHD), le clofibrate (400 mg / kg / jour; 2 fois la dose humaine) et gemfibrozil (250 mg / kg / jour;2 surface). Le fénofibrate a augmenté les adénomes d'acinars pancréatiques chez les deux sexes. Le clofibrate a augmenté le carcinome hépatocellulaire et les adénomes d'acinars pancréatiques chez les mâles et les nodules néoplasiques hépatiques chez les femelles. Le gemfibrozil a augmenté les nodules néoplasiques hépatiques chez les mâles et les femelles, tandis que les trois médicaments ont augmenté les tumeurs des cellules interstitielles testiculaires chez les mâles.
Dans une étude de 21 mois chez des souris CF-1, le fénofibrate 10, 45 et 200 mg / kg / jour (environ 0,2, 1 et 3 fois le MRHD sur la base de mg / m2 surface) a augmenté de manière significative les carcinomes hépatiques chez les deux sexes à 3 fois le MRHD. Dans une deuxième étude de 18 mois à 10, 60 et 200 mg / kg / jour, le fénofibrate a augmenté de manière significative les carcinomes hépatiques chez les souris mâles et les adénomes hépatiques chez les souris femelles à 3 fois le MRHD
Des études de microscopie électronique ont démontré une prolifération peroxisomale après administration de fénofibrate au rat. Une étude adéquate pour tester la prolifération des peroxysomes chez l'homme n'a pas été réalisée, mais des changements dans la morphologie et le nombre de peroxysomes ont été observés chez l'homme après un traitement avec d'autres membres de la classe des fibrates lorsque des biopsies hépatiques ont été comparées avant et après le traitement chez le même individu.
Mutagenèse
Il a été démontré que le fénofibrate est dépourvu de potentiel mutagène dans les tests suivants: ames, lymphome de souris, aberration chromosomique et synthèse imprévue d'ADN dans les hépatocytes primaires de rat.
Insuffisance de la fertilité
Dans les études de fertilité, les rats ont reçu des doses alimentaires orales de fénofibrate, les mâles ont reçu 61 jours avant l'accouplement et les femelles 15 jours avant l'accouplement par sevrage, ce qui n'a entraîné aucun effet indésirable sur la fertilité à des doses allant jusqu'à 300 mg / kg / jour (~ 10 fois le MRHD, basé sur mg / m2 comparaisons de surface).
Utilisation dans des populations spécifiques
Grossesse
Catégorie de grossesse C
La sécurité des femmes enceintes n'a pas été établie. Il n'y a pas d'études adéquates et bien contrôlées sur le fénofibrate chez la femme enceinte. Le fénofibrate ne doit être utilisé pendant la grossesse que si le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Chez les rats femelles ayant reçu des doses alimentaires orales de 15, 75 et 300 mg / kg / jour de fénofibrate à partir de 15 jours avant l'accouplement par sevrage, une toxicité maternelle a été observée à 0,3 fois le MRHD, sur la base de comparaisons de surface corporelle; mg / m2.
Chez les rates gravides ayant reçu des doses alimentaires orales de 14, 127 et 361 mg / kg / jour du jour de gestation 6-15 pendant la période d'organogenèse, aucun résultat de développement indésirable n'a été observé à 14 mg / kg / jour (moins de 1 fois le MRHD, basé sur des comparaisons de la surface corporelle; mg / m2). À des multiples plus élevés de doses humaines, des signes de toxicité maternelle ont été observés.
Chez les lapines gravides ayant reçu des doses de gavage oral de 15, 150 et 300 mg / kg / jour à partir du jour de gestation 618 pendant la période d'organogenèse et autorisées à délivrer, des portées avortées ont été observées à 150 mg / kg / jour (10 fois le MRHD, basé sur des comparaisons de surface corporelle: mg / m2). Aucun résultat sur le développement n'a été observé à 15 mg / kg / jour (à moins de 1 fois le MRHD, sur la base de comparaisons de surface corporelle; mg / m2).
Chez les rates gravides ayant reçu des doses alimentaires orales de 15, 75 et 300 mg / kg / jour du jour de gestation 15 au jour de lactation 21 (sevrage), une toxicité maternelle a été observée à moins de 1 fois la dose humaine maximale recommandée (MRHD), sur la base de comparaisons de la surface corporelle; mg / m2.
Mères infirmières
Le fénofibrate ne doit pas être utilisé chez les mères allaitantes. Il convient de décider d'interrompre l'allaitement ou d'interrompre le médicament, en tenant compte de l'importance du médicament pour la mère.
Utilisation pédiatrique
La sécurité et l'efficacité n'ont pas été établies chez les patients pédiatriques.
Utilisation gériatrique
L'acide fénofibrique est connu pour être considérablement excrété par le rein, et le risque d'effets indésirables de ce médicament peut être plus élevé chez les patients présentant une insuffisance rénale. L'exposition à l'acide fénofibrique n'est pas influencée par l'âge. Étant donné que les patients âgés ont une incidence plus élevée d'insuffisance rénale, la sélection des doses pour les personnes âgées doit être effectuée en fonction de la fonction rénale. Les patients âgés ayant une fonction rénale normale ne doivent pas nécessiter de modification de la dose. Envisagez de surveiller la fonction rénale chez les patients âgés prenant TRICOR
Insuffisance rénale
L'utilisation de TRICOR doit être évitée chez les patients présentant une insuffisance rénale sévère. Une réduction de la dose est nécessaire chez les patients présentant une insuffisance rénale légère à modérée. La surveillance de la fonction rénale chez les patients atteints d'insuffisance rénale est recommandée.
Insuffisance hépatique
L'utilisation de TRICOR n'a pas été évaluée chez les sujets atteints d'insuffisance hépatique.
EFFETS CÔTÉ
Expérience des essais cliniques
Étant donné que les études cliniques sont menées dans des conditions très variables, les taux d'effets indésirables observés dans les études cliniques d'un médicament ne peuvent pas être directement comparés aux taux dans les études cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
Les événements indésirables rapportés par 2% ou plus des patients traités par fénofibrate (et supérieur au placebo) au cours des essais contrôlés contre placebo en double aveugle, quelle que soit la causalité, sont répertoriés dans le tableau 1 ci-dessous. Des événements indésirables ont conduit à l'arrêt du traitement chez 5,0% des patients traités par fénofibrate et 3,0% traités par placebo. L'augmentation des tests de la fonction hépatique a été l'événement le plus fréquent, provoquant l'arrêt du traitement par fénofibrate chez 1,6% des patients dans des essais en double aveugle.
Tableau 1. Effets indésirables signalés par 2% ou plus de patients traités par Fénofibrate et supérieurs à Placebo lors des essais contrôlés par placebo en double aveugle
| SYSTÈME CORPOREL Réaction indésirable |
Fénofibrate * | Placebo |
| (N = 439) | (N = 439) | |
| CORPS ENTIER | ||
| Douleur abdominale | 4,6% | 4,4% |
| Douleur au dos | 3,4% | 2,5% |
| Maux de tête | 3,2% | 2,7% |
| DIGESTIVE | ||
| Nausées | 2,3% | 1,9% |
| Constipation | 2,1% | 1,4% |
| TROUBLES MÉTABOLIQUES ET NUTRITIONNELS | ||
| Tests anormaux de la fonction hépatique | 7,5% ** | 1,4% |
| ALT accru | 3,0% | 1,6% |
| CPK accru | 3,0% | 1,4% |
| AST accru | 3,4% ** | 0,5% |
| RESPIRATOIRE | ||
| Trouble respiratoire | 6,2% | 5,5% |
| Rhinite | 2,3% | 1,1% |
| * Dosage équivalent à 160 mg TRICOR . ** Significativement différent de Placebo. |
||
Expérience post-commercialisation
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation du fénofibrate: myalgie, rhabdomyolyse, pancréatite, insuffisance rénale aiguë, spasme musculaire, hépatite, cirrhose, anémie, arthralgie, diminution de l'hémoglobine, diminution de l'hématocrite, diminution des globules blancs, asthénie et niveaux de cholestérol HDL gravement déprimés. Étant donné que ces réactions sont rapportées volontairement à partir d'une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament.
INTERACTIONS DE DROGUES
Coumarin Anticoagulants
Une potentialisation des effets anticoagulants de type coumarine a été observée avec une prolongation du PT / INR
La prudence s'impose lorsque des anticoagulants coumarins sont administrés en association avec TRICOR. La posologie des anticoagulants doit être réduite pour maintenir le PT / INR au niveau souhaité afin de prévenir les complications hémorragiques. Des déterminations PT / INR fréquentes sont recommandées jusqu'à ce qu'il soit définitivement déterminé que le PT / INR s'est stabilisé.
Immunosuppresseurs
Les immunosuppresseurs tels que la cyclosporine et le tacrolimus peuvent produire une néphrotoxicité avec des diminutions de la clairance de la créatinine et des augmentations de la créatinine sérique, et parce que l'excrétion rénale est la principale voie d'élimination des médicaments contre le fibrate, y compris TRICOR, il existe un risque qu'une interaction conduise à une détérioration de la fonction rénale. Les avantages et les risques de l'utilisation de TRICOR (comprimés de fénofibrate) avec des immunosuppresseurs et d'autres agents potentiellement néphrotoxiques doivent être soigneusement pris en compte, et la dose efficace la plus faible utilisée et la fonction rénale surveillée.
Résines de liaison à l'acide biliaire
Étant donné que les résines de liaison à l'acide biliaire peuvent se lier à d'autres médicaments administrés simultanément, les patients doivent prendre TRICOR au moins 1 heure avant ou 4 à 6 heures après une résine de liaison à l'acide biliaire pour éviter d'entraver son absorption.
Colchicine
Des cas de myopathie, y compris une rhabdomyolyse, ont été rapportés avec des fénofibrates co-administrés avec de la colchicine, et la prudence est de mise lors de la prescription de fénofibrate avec de la colchicine.
Catégorie de grossesse C
La sécurité des femmes enceintes n'a pas été établie. Il n'y a pas d'études adéquates et bien contrôlées sur le fénofibrate chez la femme enceinte. Le fénofibrate ne doit être utilisé pendant la grossesse que si le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Chez les rats femelles ayant reçu des doses alimentaires orales de 15, 75 et 300 mg / kg / jour de fénofibrate à partir de 15 jours avant l'accouplement par sevrage, une toxicité maternelle a été observée à 0,3 fois le MRHD, sur la base de comparaisons de surface corporelle; mg / m2.
Chez les rates gravides ayant reçu des doses alimentaires orales de 14, 127 et 361 mg / kg / jour du jour de gestation 6-15 pendant la période d'organogenèse, aucun résultat de développement indésirable n'a été observé à 14 mg / kg / jour (moins de 1 fois le MRHD, basé sur des comparaisons de la surface corporelle; mg / m2). À des multiples plus élevés de doses humaines, des signes de toxicité maternelle ont été observés.
Chez les lapines gravides ayant reçu des doses de gavage oral de 15, 150 et 300 mg / kg / jour à partir du jour de gestation 618 pendant la période d'organogenèse et autorisées à délivrer, des portées avortées ont été observées à 150 mg / kg / jour (10 fois le MRHD, basé sur des comparaisons de surface corporelle: mg / m2). Aucun résultat sur le développement n'a été observé à 15 mg / kg / jour (à moins de 1 fois le MRHD, sur la base de comparaisons de surface corporelle; mg / m2).
Chez les rates gravides ayant reçu des doses alimentaires orales de 15, 75 et 300 mg / kg / jour du jour de gestation 15 au jour de lactation 21 (sevrage), une toxicité maternelle a été observée à moins de 1 fois la dose humaine maximale recommandée (MRHD), sur la base de comparaisons de la surface corporelle; mg / m2.
Expérience des essais cliniques
Étant donné que les études cliniques sont menées dans des conditions très variables, les taux d'effets indésirables observés dans les études cliniques d'un médicament ne peuvent pas être directement comparés aux taux dans les études cliniques d'un autre médicament et peuvent ne pas refléter les taux observés dans la pratique.
Les événements indésirables rapportés par 2% ou plus des patients traités par fénofibrate (et supérieur au placebo) au cours des essais contrôlés contre placebo en double aveugle, quelle que soit la causalité, sont répertoriés dans le tableau 1 ci-dessous. Des événements indésirables ont conduit à l'arrêt du traitement chez 5,0% des patients traités par fénofibrate et 3,0% traités par placebo. L'augmentation des tests de la fonction hépatique a été l'événement le plus fréquent, provoquant l'arrêt du traitement par fénofibrate chez 1,6% des patients dans des essais en double aveugle.
Tableau 1. Effets indésirables signalés par 2% ou plus de patients traités par Fénofibrate et supérieurs à Placebo lors des essais contrôlés par placebo en double aveugle
| SYSTÈME CORPOREL Réaction indésirable |
Fénofibrate * | Placebo |
| (N = 439) | (N = 439) | |
| CORPS ENTIER | ||
| Douleur abdominale | 4,6% | 4,4% |
| Douleur au dos | 3,4% | 2,5% |
| Maux de tête | 3,2% | 2,7% |
| DIGESTIVE | ||
| Nausées | 2,3% | 1,9% |
| Constipation | 2,1% | 1,4% |
| TROUBLES MÉTABOLIQUES ET NUTRITIONNELS | ||
| Tests anormaux de la fonction hépatique | 7,5% ** | 1,4% |
| ALT accru | 3,0% | 1,6% |
| CPK accru | 3,0% | 1,4% |
| AST accru | 3,4% ** | 0,5% |
| RESPIRATOIRE | ||
| Trouble respiratoire | 6,2% | 5,5% |
| Rhinite | 2,3% | 1,1% |
| * Dosage équivalent à 160 mg TRICOR . ** Significativement différent de Placebo. |
||
Expérience post-commercialisation
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation du fénofibrate: myalgie, rhabdomyolyse, pancréatite, insuffisance rénale aiguë, spasme musculaire, hépatite, cirrhose, anémie, arthralgie, diminution de l'hémoglobine, diminution de l'hématocrite, diminution des globules blancs, asthénie et niveaux de cholestérol HDL gravement déprimés. Étant donné que ces réactions sont rapportées volontairement à partir d'une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale avec l'exposition au médicament.
Il n'y a pas de traitement spécifique pour un surdosage avec TRICOR. Des soins de soutien généraux au patient sont indiqués, y compris la surveillance des signes vitaux et l'observation de l'état clinique, en cas de surdosage. Si cela est indiqué, l'élimination du médicament non absorbé doit être obtenue par vomissement ou lavage gastrique; des précautions habituelles doivent être observées pour maintenir les voies respiratoires. L'acide fénofibrique étant fortement lié aux protéines plasmatiques, l'hémodialyse ne doit pas être envisagée.
Diverses études cliniques ont démontré que des niveaux élevés de C total, de LDL-C et d'apo B, un complexe de membranes LDL, sont associés à l'athérosclérose humaine. De même, des niveaux réduits de HDL-C et de son complexe de transport, l'apolipoprotéine A (apo AI et apo AII) sont associés avec le développement de l'athérosclérose. Les investigations épidémiologiques ont établi que la morbidité et la mortalité cardiovasculaires varient directement avec le niveau de C total, LDL-C et TG, et inversement avec le niveau de HDL-C. L'effet indépendant de l'augmentation du HDL-C ou de la réduction des triglycérides (TG) sur le risque de morbidité et de mortalité cardiovasculaires n'a pas été déterminé.
L'acide fénofibrique, le métabolite actif du fénofibrate, produit des réductions du cholestérol total, du cholestérol LDL, de l'apolipoprotéine B, des triglycérides totaux et des lipoprotéines riches en triglycérides (VLDL) chez les patients traités. De plus, le traitement par fénofibrate entraîne une augmentation de la lipoprotéine haute densité (HDL) et des apolipoprotéines apoAI et apoAII
Le fénofibrate est un promédicament de l'acide fénofibrique à fraction chimique active. Le fénofibrate est converti par hydrolyse ester dans le corps en acide fénofibrique qui est le constituant actif mesurable dans la circulation.
Absorption
La biodisponibilité absolue du fénofibrate ne peut pas être déterminée car le composé est pratiquement insoluble dans les milieux aqueux adaptés à l'injection. Cependant, le fénofibrate est bien absorbé par le tractus gastro-intestinal. Après administration orale chez des volontaires sains, environ 60% d'une dose unique de fénofibrate radiomarqué est apparue dans l'urine, principalement sous forme d'acide fénofibrique et de son conjugué glucuronate, et 25% a été excrétée dans les fèces. Les concentrations plasmatiques maximales d'acide fénofibrique surviennent dans les 6 à 8 heures suivant l'administration.
L'absorption du fénofibrate est augmentée lorsqu'il est administré avec de la nourriture. Avec les comprimés de fénofibrate, le degré d'absorption est augmenté d'environ 35% sous alimentation par rapport aux conditions de jeûne.
Distribution
Lors de doses multiples de fénofibrate, l'état d'équilibre de l'acide fénofibrique est atteint en 5 jours. Les concentrations plasmatiques d'acide fénofibrique à l'état d'équilibre sont environ le double de celles qui suivent une dose unique. La liaison aux protéines sériques était d'environ 99% chez les sujets normaux et hyperlipidémiques.
Métabolisme
Après administration orale, le fénofibrate est rapidement hydrolysé par les estérases en métabolite actif, l'acide fénofibrique; aucun fénofibrate inchangé n'est détecté dans le plasma.
L'acide fénofibrique est principalement conjugué à l'acide glucuronique puis excrété dans l'urine. Une petite quantité d'acide fénofibrique est réduite au niveau de la fraction carbonyle en un métabolite benzhydrol qui est, à son tour, conjugué à l'acide glucuronique et excrété dans l'urine.
In vivo les données du métabolisme indiquent que ni le fénofibrate ni l'acide fénofibrique ne subissent de métabolisme oxydatif (par ex., cytochrome P450) dans une large mesure.
Élimination
Après absorption, le fénofibrate est principalement excrété dans l'urine sous forme de métabolites, principalement l'acide fénofibrique et le glucuronide d'acide fénofibrique. Après administration de fénofibrate radiomarqué, environ 60% de la dose est apparue dans l'urine et 25% a été excrétée dans les fèces.
L'acide fénofibrique est éliminé avec une demi-vie de 20 heures, permettant un dosage une fois par jour.
However, we will provide data for each active ingredient





