


Komposition:
Anwendung:
Wird bei der Behandlung verwendet:
Medizinisch geprüft von Oliinyk Elizabeth Ivanovna, Apotheke Zuletzt aktualisiert am 10.04.2022

Achtung! Die Informationen auf der Seite sind nur für medizinisches Fachpersonal! Die Informationen werden in öffentlichen Quellen gesammelt und können aussagekräftige Fehler enthalten! Seien Sie vorsichtig und überprüfen Sie alle Informationen auf dieser Seite!
Dosierungsformen und Stärken
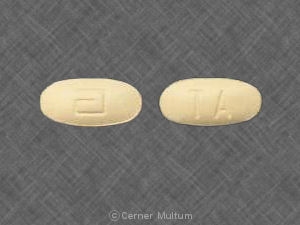
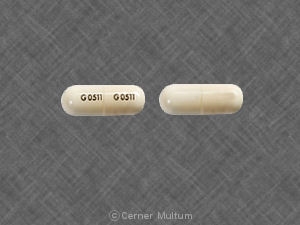
- 54 mg gelbe Tabletten, bedruckt mit dem Logo „a“ und den Codeidentifikationsbuchstaben "TA".
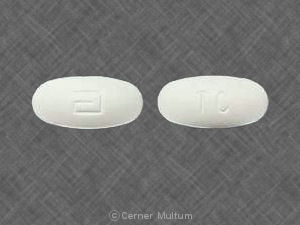
- 160 mg weiße Tabletten, bedruckt mit dem Logo „a“ und den Codeidentifikationsbuchstaben "TC".
Lagerung und Handhabung
TRICOR® (Fenofibrat-Tabletten) ist in zwei Stärken erhältlich:
54 mg gelbe Tabletten, bedruckt mit dem Logo „a“ und den Codeidentifikationsbuchstaben "TA", erhältlich in Flaschen mit 90 Stück (NDC 0074-4009-90).
160 mg weiße Tabletten, bedruckt mit dem Logo „a“ und den Codeidentifikationsbuchstaben "TC", erhältlich in Flaschen mit 90 Stück (NDC 0074-4013-90).
Lagerung
Bei 25 ° C lagern; Ausflüge bis 15-30 ° C (59-86 ° F) erlaubt.
Darf nicht in die Hände von Kindern gelangen. Vor Feuchtigkeit schützen.
Hergestellt von: Laboratoires Fournier, S. A., 21300 Chen´ve, Frankreich. Überarbeitet: Januar 2017.
Primäre Hypercholesterinämie oder gemischte Dyslipidämie
TRICOR ist als Zusatztherapie zur Ernährung angezeigt, um das erhöhte Lipoproteincholesterin niedriger Dichte (LDL-C), das Gesamtcholesterin (Total-C), Triglyceride und Apolipoprotein B (Apo B) zu senken und das Lipoproteincholesterin hoher Dichte (HDL-C) zu erhöhen ) bei erwachsenen Patienten mit primärer Hypercholesterinämie oder gemischter Dyslipidämie.
Schwere Hypertriglyceridämie
TRICOR ist auch als Zusatztherapie zur Ernährung zur Behandlung von erwachsenen Patienten mit schwerer Hypertriglyceridämie angezeigt. Eine Verbesserung der Blutzuckerkontrolle bei Diabetikern, die eine Fastenchylomikronämie zeigen, vermeidet normalerweise die Notwendigkeit einer pharmakologischen Intervention.
Deutlich erhöhte Serumtriglyceridspiegel (z. > 2.000 mg / dl) können das Risiko einer Pankreatitis erhöhen. Die Wirkung der Fenofibrat-Therapie auf die Verringerung dieses Risikos wurde nicht ausreichend untersucht.
Wichtige Nutzungsbeschränkungen
In einer großen, randomisierten kontrollierten Studie mit Patienten mit Typ-2-Diabetes mellitus wurde nicht gezeigt, dass Fenofibrat in einer Dosis von 160 mg TRICOR die Morbidität und Mortalität koronarer Herzkrankheiten verringert.
Allgemeine Überlegungen
Die Patienten sollten vor der Einnahme von TRICOR eine geeignete lipidsenkende Diät erhalten und diese Diät während der Behandlung mit TRICOR fortsetzen. TRICOR-Tabletten sollten zu den Mahlzeiten verabreicht werden, wodurch die Bioverfügbarkeit des Medikaments optimiert wird.
Die anfängliche Behandlung von Dyslipidämie ist eine diätetische Therapie, die für die Art der Lipoproteinanomalie spezifisch ist. Übermäßiges Körpergewicht und übermäßiger Alkoholkonsum können wichtige Faktoren bei Hypertriglyceridämie sein und sollten vor jeder medikamentösen Therapie angegangen werden. Körperliche Bewegung kann eine wichtige Nebenmaßnahme sein. Krankheiten, die zu Hyperlipidämie beitragen, wie Hypothyreose oder Diabetes mellitus, sollten gesucht und angemessen behandelt werden. Östrogentherapie, Thiaziddiuretika und Betablocker sind manchmal mit einem massiven Anstieg der Plasmatriglyceride verbunden, insbesondere bei Patienten mit familiärer Hypertriglyceridämie. In solchen Fällen kann das Absetzen des spezifischen ätiologischen Mittels die Notwendigkeit einer spezifischen medikamentösen Therapie der Hypertriglyceridämie vermeiden.
Die Lipidspiegel sollten regelmäßig überwacht werden, und es sollte erwogen werden, die TRICOR-Dosis zu reduzieren, wenn die Lipidspiegel deutlich unter den Zielbereich fallen.
Die Therapie sollte bei Patienten abgebrochen werden, die nach zweimonatiger Behandlung mit der empfohlenen Höchstdosis von 160 mg einmal täglich kein angemessenes Ansprechen haben.
Primäre Hypercholesterinämie oder gemischte Dyslipidämie
Die Anfangsdosis von TRICOR beträgt 160 mg einmal täglich.
Schwere Hypertriglyceridämie
Die Anfangsdosis beträgt 54 bis 160 mg pro Tag. Die Dosierung sollte entsprechend der Reaktion des Patienten individualisiert und gegebenenfalls nach wiederholten Lipidbestimmungen in Intervallen von 4 bis 8 Wochen angepasst werden. Die maximale Dosis beträgt 160 mg einmal täglich.
Beeinträchtigte Nierenfunktion
Die Behandlung mit TRICOR sollte bei Patienten mit leichter bis mäßig beeinträchtigter Nierenfunktion in einer Dosis von 54 mg pro Tag begonnen und erst nach Bewertung der Auswirkungen auf die Nierenfunktion und die Lipidspiegel bei dieser Dosis erhöht werden. Die Anwendung von TRICOR sollte bei Patienten mit schwerer Nierenfunktionsstörung vermieden werden.
Geriatrische Patienten
Die Dosisauswahl für ältere Menschen sollte auf der Grundlage der Nierenfunktion erfolgen.
TRICOR ist kontraindiziert in:
- Patienten mit schwerer Nierenfunktionsstörung, einschließlich Patienten, die eine Dialyse erhalten.
- Patienten mit aktiver Lebererkrankung, einschließlich Patienten mit primärer biliärer Zirrhose und ungeklärte anhaltende Leberfunktionsstörungen.
- Patienten mit bereits bestehender Gallenblasenerkrankung.
- stillende Mütter
- Patienten mit bekannter Überempfindlichkeit gegen Fenofibrat oder Fenofibrinsäure.
WARNHINWEISE
Im Rahmen der enthalten "PRECAUTIONS" Sektion
VORSICHTSMASSNAHMEN
Sterblichkeit und Morbidität der koronaren Herzkrankheit
Die Wirkung von TRICOR auf die Morbidität und Mortalität von koronaren Herzerkrankungen sowie auf die nicht kardiovaskuläre Mortalität wurde nicht nachgewiesen.
Die Studie zur Bekämpfung des kardiovaskulären Risikos bei Diabetes Lipid (ACCORD Lipid) war eine randomisierte placebokontrollierte Studie an 5518 Patienten mit Typ-2-Diabetes mellitus unter mit Fenofibrat behandelter Hintergrundstatintherapie. Die mittlere Follow-up-Dauer betrug 4,7 Jahre. Die Kombinationstherapie mit Fenofibrat plus Statin zeigte eine nicht signifikante relative Risikoreduktion von 8% im primären Ergebnis schwerwiegender unerwünschter kardiovaskulärer Ereignisse (MACE), die aus nicht tödlichem Myokardinfarkt, nicht tödlichem Schlaganfall und Tod von Herz-Kreislauf-Erkrankungen besteht (Hazard Ratio [HR] 0,92, 95% CI 0,79-1,0,0,3 bis (im Vergleich zu Mon2) (p (p). In einer Gender-Untergruppenanalyse betrug die Hazard Ratio für MACE bei Männern, die eine Kombinationstherapie erhielten, gegenüber einer Statin-Monotherapie 0,82 (95% CI 0,69-0,99), und die Hazard Ratio für MACE bei Frauen, die eine Kombinationstherapie erhalten, betrug 1,38 (95% CI 0,98-1,94) (Interaktion p = 001). Die klinische Bedeutung dieses Untergruppenfundes ist unklar.
Die FIELD-Studie (Fenofibrat Intervention and Event Lowering in Diabetes) war eine 5-jährige randomisierte, placebokontrollierte Studie an 9795 Patienten mit Typ-2-Diabetes mellitus, die mit Fenofibrat behandelt wurden. Fenofibrat zeigte eine nicht signifikante relative Verringerung des primären Ergebnisses von Ereignissen bei koronaren Herzerkrankungen um 11% (Hazard Ratio [HR] 0,89, 95% CI 0,75-1,05, p = 0,16) und eine signifikante Verringerung des sekundären Ergebnisses von 11% Gesamtereignisse bei Herz-Kreislauf-Erkrankungen (HR 0,89,89 [0,80,80,80). Es gab einen nicht signifikanten Anstieg der Gesamt- bzw. Koronarherzkrankheitssterblichkeit um 11% (HR 1,11 [0,95, 1,29], p = 0,18) und 19% (HR 1,19 [0,90, 1,57], p = 0,22) mit Fenofibrat im Vergleich zu Placebo.
Aufgrund chemischer, pharmakologischer und klinischer Ähnlichkeiten zwischen TRICOR (Fenofibrat-Tabletten), Clofibrat und Gemfibrozil können die nachteiligen Befunde in 4 großen randomisierten, placebokontrollierten klinischen Studien mit diesen anderen Fibrat-Medikamenten auch für TRICOR gelten
Im Coronary Drug Project, einer großen Studie zum postmyokardialen Infarkt von Patienten, die 5 Jahre lang mit Clofibrat behandelt wurden, wurde kein Unterschied in der Mortalität zwischen der Clofibrat-Gruppe und der Placebo-Gruppe festgestellt. Es gab jedoch einen Unterschied in der Geschwindigkeit der Cholelithiasis und Cholezystitis, die eine Operation zwischen den beiden Gruppen erforderten (3,0% vs. 1,8%).
In einer von der Weltgesundheitsorganisation (WHO) durchgeführten Studie wurden 5000 Probanden ohne bekannte Erkrankung der Koronararterien 5 Jahre lang mit Placebo oder Clofibrat behandelt und weitere ein Jahr lang beobachtet. In der Clofibrat-Gruppe gab es im Vergleich zur Placebo-Gruppe eine statistisch signifikante, altersgerechte Gesamtmortalität (5,70% vs. 3,96%, p = <0,01). Eine übermäßige Mortalität war auf einen Anstieg der nicht kardiovaskulären Ursachen um 33% zurückzuführen, einschließlich Malignität, Komplikationen nach der Cholezystektomie und Pankreatitis. Dies schien das höhere Risiko einer Gallenblasenerkrankung bei mit Clofibrat behandelten Patienten zu bestätigen, die im Coronary Drug Project untersucht wurden.
Die Helsinki-Herzstudie war eine große (n = 4081) Studie an Männern mittleren Alters ohne koronare Arterienerkrankung in der Vorgeschichte. Die Probanden erhielten 5 Jahre lang entweder Placebo oder Gemfibrozil mit einer offenen Verlängerung von 3,5 Jahren. Die Gesamtmortalität war in der Gemfibrozil-Randomisierungsgruppe zahlenmäßig höher, erreichte jedoch keine statistische Signifikanz (p = 0,19, 95% -Konfidenzintervall für das relative Risiko G: P = 0,91-1,64). Obwohl die Krebstoten in der Gemfibrozil-Gruppe (p = 0,11) im Trend lagen, wurde bei Krebs (ohne Basalzellkarzinom) in beiden Studiengruppen die gleiche Häufigkeit diagnostiziert. Aufgrund der begrenzten Größe der Studie wurde nicht gezeigt, dass sich das relative Todesrisiko aus irgendeinem Grund von dem in den 9-Jahres-Follow-up-Daten aus der Studie der Weltgesundheitsorganisation (RR = 1,29) beobachteten unterscheidet.
Eine sekundäre Präventionskomponente der Helsinki-Herzstudie umfasste Männer mittleren Alters, die wegen bekannter oder vermuteter koronarer Herzkrankheit von der primären Präventionsstudie ausgeschlossen waren. Die Probanden erhielten 5 Jahre lang Gemfibrozil oder Placebo. Obwohl die Herztodesfälle in der Gemfibrozil-Gruppe tendenziell höher waren, war dies statistisch nicht signifikant (Hazard Ratio 2,2, 95% -Konfidenzintervall: 0,94-5,05). Die Rate der Gallenblasenoperationen war zwischen den Studiengruppen statistisch nicht signifikant, stieg jedoch in der Gemfibrozil-Gruppe (1,9% vs. 0,3%, p = 0,07).
Skelettmuskel
Fibrate erhöhen das Risiko für Myopathie und wurden mit Rhabdomyolyse in Verbindung gebracht. Das Risiko für eine schwerwiegende Muskeltoxizität scheint bei älteren Patienten und bei Patienten mit Diabetes, Niereninsuffizienz oder Hypothyreose erhöht zu sein.
Myopathie sollte bei jedem Patienten mit diffusen Myalgien, Muskelempfindlichkeit oder -schwäche und / oder deutlichen Erhöhungen der Kreatinphosphokinase (CPK) -Spiegel in Betracht gezogen werden.
Patienten sollten angewiesen werden, unerklärliche Muskelschmerzen, Empfindlichkeit oder Schwäche unverzüglich zu melden, insbesondere wenn sie von Unwohlsein oder Fieber begleitet werden. Die CPK-Spiegel sollten bei Patienten bewertet werden, die über diese Symptome berichten, und die TRICOR-Therapie sollte abgebrochen werden, wenn deutlich erhöhte CPK-Spiegel auftreten oder Myopathie / Myositis vermutet oder diagnostiziert wird.
Daten aus Beobachtungsstudien zeigen, dass das Risiko für Rhabdomyolyse erhöht ist, wenn Fibrate, insbesondere Gemfibrozil, zusammen mit einem HMG-CoA-Reduktase-Inhibitor (Statin) verabreicht werden. Die Kombination sollte vermieden werden, es sei denn, der Nutzen weiterer Veränderungen der Lipidspiegel überwiegt wahrscheinlich das erhöhte Risiko dieser Arzneimittelkombination.
Fälle von Myopathie, einschließlich Rhabdomyolyse, wurden mit Fenofibraten berichtet, die zusammen mit Colchicin verabreicht wurden, und bei der Verschreibung von Fenofibrat mit Colchicin ist Vorsicht geboten.
Leberfunktion
Fenofibrat in Dosen von 107 mg bis 160 mg TRICOR pro Tag wurde mit einem Anstieg der Serumtransaminasen [AST (SGOT) oder ALT (SGPT)] in Verbindung gebracht. In einer gepoolten Analyse von 10 placebokontrollierten Studien trat bei 5,3% der Patienten, die Fenofibrat einnahmen, ein Anstieg auf> das Dreifache der Obergrenze des Normalwerts auf, gegenüber 1,1% der mit Placebo behandelten Patienten.
Wenn Transaminase-Bestimmungen entweder nach Absetzen der Behandlung oder während der fortgesetzten Behandlung durchgeführt wurden, wurde normalerweise eine Rückkehr zu normalen Grenzen beobachtet. Die Inzidenz von Transaminasenanstiegen im Zusammenhang mit der Fenofibrat-Therapie scheint dosisabhängig zu sein. In einer 8-wöchigen dosisabhängigen Studie, Die Inzidenz von ALT- oder AST-Erhöhungen auf mindestens das Dreifache der Obergrenze des Normalwerts betrug 13% bei Patienten, die Dosierungen erhielten, die 107 mg bis 160 mg TRICOR pro Tag entsprachen, und betrug 0% bei Patienten, die Dosierungen erhielten, die 54 mg oder weniger TRICOR pro Tag entsprachen Tag, oder Placebo. Hepatozelluläre, chronisch aktive und cholestatische Hepatitis im Zusammenhang mit der Fenofibrat-Therapie wurde nach wochen- bis mehrjähriger Exposition berichtet. In äußerst seltenen Fällen wurde über eine Zirrhose im Zusammenhang mit einer chronisch aktiven Hepatitis berichtet.
Die Basis- und regelmäßige regelmäßige regelmäßige Überwachung der Leberfunktion, einschließlich Serum-ALT (SGPT), sollte für die Dauer der Therapie mit TRICOR durchgeführt und die Therapie abgebrochen werden, wenn die Enzymwerte über dem Dreifachen der normalen Grenze bleiben.
Serum Kreatinin
Bei Patienten mit Fenofibrat wurde über Erhöhungen des Serumkreatinins berichtet. Diese Erhöhungen kehren nach Absetzen von Fenofibrat tendenziell zum Ausgangswert zurück. Die klinische Bedeutung dieser Beobachtungen ist unbekannt. Überwachung der Nierenfunktion bei Patienten mit Nierenfunktionsstörung, die TRICOR einnehmen. Die Überwachung der Nieren sollte auch bei Patienten in Betracht gezogen werden, die TRICOR einnehmen und ein Risiko für Niereninsuffizienz haben, wie z. B. ältere Menschen und Patienten mit Diabetes.
Cholelithiasis
Fenofibrat kann wie Clofibrat und Gemfibrozil die Cholesterinausscheidung in die Galle erhöhen und zu Cholelithiasis führen. Bei Verdacht auf Cholelithiasis sind Gallenblasstudien angezeigt. Die TRICOR-Therapie sollte abgebrochen werden, wenn Gallensteine gefunden werden.
Cumarin Antikoagulanzien
Vorsicht ist geboten, wenn Cumarin-Antikoagulanzien in Verbindung mit TRICOR verabreicht werden, da die Antikoagulanzien vom Cumarin-Typ bei der Verlängerung der Prothrombin-Zeit / des International Normalized Ratio (PT / INR) wirksam sind. Um Blutungskomplikationen vorzubeugen, wird eine häufige Überwachung von PT / INR und eine Dosisanpassung des Antikoagulans empfohlen, bis sich PT / INR stabilisiert hat.
Pankreatitis
Bei Patienten, die Fenofibrat, Gemfibrozil und Clofibrat einnehmen, wurde über Pankreatitis berichtet. Dieses Auftreten kann bei Patienten mit schwerer Hypertriglyceridämie, direkter Arzneimittelwirkung oder einem sekundären Phänomen, das durch die Bildung von Gallenwegen oder Schlamm mit Verstopfung des gemeinsamen Gallengangs vermittelt wird, ein Versagen der Wirksamkeit darstellen.
Hämatologische Veränderungen
Bei Patienten nach Beginn der Fenofibrat-Therapie wurde ein leichter bis mittelschwerer Hämoglobin-, Hämatokrit- und weißer Blutkörperchenabfall beobachtet. Diese Werte stabilisieren sich jedoch während der Langzeitverabreichung. Thrombozytopenie und Agranulozytose wurden bei mit Fenofibrat behandelten Personen berichtet. Während der ersten 12 Monate der Verabreichung von TRICOR wird eine regelmäßige Überwachung der Anzahl roter und weißer Blutkörperchen empfohlen.
Überempfindlichkeitsreaktionen
Akute Überempfindlichkeitsreaktionen wie das Stevens-Johnson-Syndrom und toxische epidermale Nekrolyse, die einen Krankenhausaufenthalt des Patienten und eine Behandlung mit Steroiden erfordern, wurden bei mit Fenofibraten behandelten Personen berichtet. Urtikaria wurde in 1.1 vs. gesehen. 0% und Hautausschlag in 1,4 vs. 0,8% der Fenofibrat- bzw. Placebo-Patienten in kontrollierten Studien.
Venothromboembolische Krankheit
In der FIELD-Studie wurden Lungenembolien (PE) und tiefe Venenthrombosen (DVT) in der mit Fenofibrat behandelten Gruppe mit höheren Raten beobachtet. Von 9.795 Patienten, die an FIELD teilnahmen, gab es 4.900 in der Placebogruppe und 4.895 in der Fenofibratgruppe. Für die TVT gab es 48 Ereignisse (1%) in der Placebogruppe und 67 (1%) in der Fenofibratgruppe (p = 0,074); und für PE gab es 32 (0,7%) Ereignisse in der Placebogruppe und 53 (1%) in der Fenofibratgruppe (p = 0,022).
Im Coronary Drug Project trat bei einem höheren Anteil der Clofibratgruppe eine bestimmte oder vermutete tödliche oder nicht tödliche Lungenembolie oder Thrombophlebitis auf als in der Placebo-Gruppe (5,2% vs. 3,3% nach fünf Jahren; p <0,01).
Paradoxe Abnahmen der HDL-Cholesterinspiegel
Nach dem Inverkehrbringen und in klinischen Studien wurde über einen starken Rückgang des HDL-Cholesterinspiegels (bis zu 2 mg / dl) bei Diabetikern und Nichtdiabetikern berichtet, die mit einer Fibrattherapie begonnen wurden. Die Abnahme von HDL-C spiegelt sich in einer Abnahme des Apolipoproteins A1 wider. Es wurde berichtet, dass diese Abnahme innerhalb von 2 Wochen bis Jahren nach Beginn der Fibrattherapie auftritt. Die HDL-C-Spiegel bleiben gedrückt, bis die Fibrattherapie zurückgezogen wurde. Das Ansprechen auf den Entzug der Fibrattherapie ist schnell und anhaltend. Die klinische Bedeutung dieser Abnahme von HDL-C ist unbekannt. Es wird empfohlen, die HDL-C-Spiegel innerhalb der ersten Monate nach Beginn der Fibrattherapie zu überprüfen. Wenn ein stark depressiver HDL-C-Spiegel festgestellt wird, sollte die Fibrattherapie abgebrochen und der HDL-C-Spiegel überwacht werden, bis er wieder zu Studienbeginn zurückgekehrt ist, und die Fibrattherapie sollte nicht erneut eingeleitet werden.
Nichtklinische Toxikologie
Karzinogenese und Mutagenese und Beeinträchtigung der Fruchtbarkeit
Zwei Kanzerogenitätsstudien über die Nahrung wurden an Ratten mit Fenofibrat durchgeführt. In der ersten 24-monatigen Studie wurde Wistar-Ratten 10, 45 und 200 mg / kg / Tag, ungefähr 0,3, 1 und 6-mal so viel wie die empfohlene maximale menschliche Dosis (MRHD), basierend auf Vergleichen der Körperoberfläche (mg / m2). Bei einer Dosis von 200 mg / kg / Tag (beim 6-fachen der MRHD) war die Inzidenz von Leberkarzinomen bei beiden Geschlechtern signifikant erhöht. Ein statistisch signifikanter Anstieg der Pankreaskarzinome wurde bei Männern beim 1- und 6-fachen der MRHD beobachtet; Bei Männern wurde ein Anstieg der Pankreasadenome und benignen testikulären interstitiellen Zelltumoren bei Männern beobachtet. In einer zweiten 24-monatigen Studie zur Kanzerogenität von Ratten bei einem anderen Rattenstamm (Sprague-Dawley) Dosen von 10 und 60 mg / kg / Tag (0,3 und 2 mal MRHD) führte bei beiden Geschlechtern zu einem signifikanten Anstieg der Inzidenz von Pankreas-Azinar-Adenomen und bei Männern zum 2-fachen der MRHD zu einem Anstieg der testikulären interstitiellen Zelltumoren
Eine 117-wöchige Kanzerogenitätsstudie wurde an Ratten durchgeführt, in der drei Arzneimittel verglichen wurden: Fenofibrat 10 und 60 mg / kg / Tag (0,3- und 2-fache MRHD), Clofibrat (400 mg / kg / Tag; 2-fache menschliche Dosis) und Gemfibrozil (250 mg / kg / Tag; / mg / mg / m. basierend auf2 Oberfläche). Fenofibrat erhöhte die Adenome von Pankreas-Azinar bei beiden Geschlechtern. Clofibrat erhöhte das hepatozelluläre Karzinom und die Pankreas-Azinar-Adenome bei Männern und die neoplastischen Leberknoten bei Frauen. Gemfibrozil erhöhte die neoplastischen Knötchen in der Leber bei Männern und Frauen, während alle drei Medikamente die testikulären interstitiellen Zelltumoren bei Männern erhöhten.
In einer 21-monatigen Studie an CF-1-Mäusen wurde Fenofibrat 10, 45 und 200 mg / kg / Tag (ungefähr 0,2, 1 und 3-mal MRHD auf Basis von mg / m)2 Oberfläche) erhöhte die Leberkarzinome bei beiden Geschlechtern beim 3-fachen der MRHD signifikant. In einer zweiten 18-monatigen Studie mit 10, 60 und 200 mg / kg / Tag erhöhte Fenofibrat die Leberkarzinome bei männlichen Mäusen und Leberadenome bei weiblichen Mäusen signifikant zum 3-fachen der MRHD
Elektronenmikroskopische Studien haben eine peroxisomale Proliferation nach Fenofibrat-Verabreichung an die Ratte gezeigt. Eine angemessene Studie zum Testen auf Peroxisomproliferation beim Menschen wurde nicht durchgeführt, aber Veränderungen der Peroxisommorphologie und -zahlen wurden beim Menschen nach Behandlung mit anderen Mitgliedern der Fibratklasse beobachtet, wenn Leberbiopsien vor und nach der Behandlung bei derselben Person verglichen wurden.
Mutagenese
In den folgenden Tests wurde gezeigt, dass Fenofibrat kein mutagenes Potential aufweist: Ames, Mauslymphom, Chromosomenaberration und außerplanmäßige DNA-Synthese in primären Rattenhepatozyten.
Beeinträchtigung der Fruchtbarkeit
In Fruchtbarkeitsstudien erhielten Ratten orale Nahrungsdosen von Fenofibrat, Männer 61 Tage vor der Paarung und Frauen 15 Tage vor der Paarung durch Entwöhnung, was bei Dosen von bis zu 300 mg / kg / Tag (~ 10-mal) keine nachteiligen Auswirkungen auf die Fruchtbarkeit hatte MRHD, basierend auf mg / m2 Oberflächenvergleiche).
Verwendung in bestimmten Populationen
Schwangerschaft
Schwangerschaftskategorie C
Die Sicherheit bei schwangeren Frauen wurde nicht nachgewiesen. Es gibt keine angemessenen und gut kontrollierten Studien zu Fenofibrat bei schwangeren Frauen. Fenofibrat sollte während der Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen das potenzielle Risiko für den Fötus rechtfertigt.
Bei weiblichen Ratten, denen 15 Tage vor der Paarung durch Absetzen orale Nahrungsdosen von 15, 75 und 300 mg / kg / Tag Fenofibrat verabreicht wurden, wurde die maternale Toxizität bei 0,3-facher MRHD beobachtet, basierend auf Vergleichen der Körperoberfläche. mg / m2.
Bei trächtigen Ratten, denen während des Zeitraums der Organogenese orale Nahrungsdosen von 14, 127 und 361 mg / kg / Tag ab dem 6-15. Schwangerschaftstag verabreicht wurden, wurden bei 14 mg / kg / Tag (weniger als das 1-fache der MRHD) keine nachteiligen Entwicklungsergebnisse beobachtet basierend auf Vergleichen der Körperoberfläche; mg / m2). Bei höheren Vielfachen menschlicher Dosen wurde ein Nachweis der maternalen Toxizität beobachtet.
Bei trächtigen Kaninchen, denen ab dem Schwangerschaftstag 618 während des Zeitraums der Organogenese orale Sondendosen von 15, 150 und 300 mg / kg / Tag verabreicht wurden und die abgegeben werden durften, wurden abgebrochene Würfe mit 150 mg / kg / Tag (10-fache MRHD) beobachtet. basierend auf Vergleichen der Körperoberfläche: mg / m2). Bei 15 mg / kg / Tag (bei weniger als dem 1-fachen der MRHD, basierend auf Vergleichen der Körperoberfläche; mg / m) wurden keine Entwicklungsergebnisse beobachtet2).
Bei trächtigen Ratten, denen vom 15. Schwangerschaftstag bis zum 21. Laktationstag (Absetzen) orale Nahrungsdosen von 15, 75 und 300 mg / kg / Tag verabreicht wurden, wurde die maternale Toxizität bei weniger als dem 1-fachen der empfohlenen maximalen menschlichen Dosis (MRHD) beobachtet auf Vergleiche der Körperoberfläche; mg / m2.
Stillende Mütter
Fenofibrat sollte nicht bei stillenden Müttern angewendet werden. Es sollte entschieden werden, ob die Krankenpflege abgebrochen oder das Medikament abgesetzt werden soll, wobei die Bedeutung des Arzneimittels für die Mutter zu berücksichtigen ist.
Pädiatrische Anwendung
Sicherheit und Wirksamkeit wurden bei pädiatrischen Patienten nicht nachgewiesen.
Geriatrische Anwendung
Es ist bekannt, dass Fenofibrinsäure im Wesentlichen über die Niere ausgeschieden wird, und das Risiko von Nebenwirkungen dieses Arzneimittels kann bei Patienten mit eingeschränkter Nierenfunktion größer sein. Die Exposition gegenüber Fenofibric-Säure wird nicht vom Alter beeinflusst. Da ältere Patienten eine höhere Inzidenz von Nierenfunktionsstörungen aufweisen, sollte die Dosisauswahl für ältere Menschen auf der Grundlage der Nierenfunktion erfolgen. Ältere Patienten mit normaler Nierenfunktion sollten keine Dosisänderungen verlangen. Erwägen Sie die Überwachung der Nierenfunktion bei älteren Patienten, die TRICOR einnehmen
Nierenfunktionsstörung
Die Anwendung von TRICOR sollte bei Patienten mit schwerer Nierenfunktionsstörung vermieden werden. Bei Patienten mit leichter bis mittelschwerer Nierenfunktionsstörung ist eine Dosisreduktion erforderlich. Die Überwachung der Nierenfunktion bei Patienten mit eingeschränkter Nierenfunktion wird empfohlen.
Leberfunktionsstörung
Die Anwendung von TRICOR wurde bei Patienten mit Leberfunktionsstörung nicht bewertet.
SEITENWIRKUNGEN
Erfahrung in klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können in den klinischen Studien eines Arzneimittels beobachtete Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien eines anderen Arzneimittels verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Unerwünschte Ereignisse, die von 2% oder mehr der mit Fenofibrat (und mehr als Placebo) behandelten Patienten während der doppelblinden, placebokontrollierten Studien unabhängig von ihrer Kausalität gemeldet wurden, sind in der folgenden Tabelle 1 aufgeführt. Unerwünschte Ereignisse führten bei 5,0% der mit Fenofibrat behandelten Patienten und bei 3,0%, die mit Placebo behandelt wurden, zum Absetzen der Behandlung. Erhöhungen der Leberfunktionstests waren die häufigsten Ereignisse, die bei 1,6% der Patienten in Doppelblindstudien zum Absetzen der Fenofibrat-Behandlung führten.
Tabelle 1. Unerwünschte Reaktionen, die von 2% oder mehr der mit Fenofibrat behandelten Patienten gemeldet wurden und während der doppelblinden, placebokontrollierten Studien größer als Placebo waren
| KÖRPERSYSTEM Unerwünschte Reaktion |
Fenofibrat * | Placebo |
| (N = 439) | (N = 439) | |
| KÖRPER ALS GANZ | ||
| Bauchschmerzen | 4,6% | 4,4% |
| Rückenschmerzen | 3,4% | 2,5% |
| Kopfschmerzen | 3,2% | 2,7% |
| DIGESTIVE | ||
| Übelkeit | 2,3% | 1,9% |
| Verstopfung | 2,1% | 1,4% |
| METABOLISCHE UND ERNÄHRUNGSWESEN | ||
| Abnormale Leberfunktionstests | 7,5% ** | 1,4% |
| Erhöhte ALT | 3,0% | 1,6% |
| Erhöhte CPK | 3,0% | 1,4% |
| Erhöhte AST | 3,4% ** | 0,5% |
| VERLETZUNG | ||
| Atemstörung | 6,2% | 5,5% |
| Rhinitis | 2,3% | 1,1% |
| * Dosierung entsprechend 160 mg TRICOR . ** Deutlich anders als Placebo. |
||
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von Fenofibrat nach der Zulassung festgestellt: Myalgie, Rhabdomyolyse, Pankreatitis, akutes Nierenversagen, Muskelkrampf, Hepatitis, Zirrhose, Anämie, Arthralgie, Abnahme des Hämoglobins, Abnahme des Hämatokrits, Abnahme der weißen Blutkörperchen, Asthenie und stark depressive HDL-Cholesterinspiegel. Da diese Reaktionen freiwillig von einer Population ungewisser Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen ursächlichen Zusammenhang mit der Arzneimittelexposition herzustellen.
Drogeninteraktionen
Cumarin Antikoagulanzien
Eine Potenzierung von Antikoagulanzien vom Cumarin-Typ wurde mit Verlängerung des PT / INR beobachtet
Vorsicht ist geboten, wenn Cumarin-Antikoagulanzien in Verbindung mit TRICOR verabreicht werden. Die Dosierung der Antikoagulanzien sollte reduziert werden, um den PT / INR auf dem gewünschten Niveau zu halten und Blutungskomplikationen zu vermeiden. Häufige PT / INR-Bestimmungen sind ratsam, bis definitiv festgestellt wurde, dass sich der PT / INR stabilisiert hat.
Immunsuppressiva
Immunsuppessiva wie Cyclosporin und Tacrolimus können eine Nephrotoxizität mit einer Abnahme der Kreatinin-Clearance und einem Anstieg des Serumkreatinins hervorrufen. Da die renale Ausscheidung der primäre Eliminationsweg für Fibratmedikamente einschließlich TRICOR ist, besteht das Risiko, dass eine Wechselwirkung zu einer Verschlechterung der Nierenfunktion führt. Der Nutzen und das Risiko der Verwendung von TRICOR (Fenofibrat-Tabletten) mit Immunsuppressiva und anderen potenziell nephrotoxischen Mitteln sollten sorgfältig abgewogen und die niedrigste wirksame Dosis und Nierenfunktion überwacht werden.
Bile Acid Binding Resins
Da Gallensäure-Bindungsharze andere gleichzeitig verabreichte Arzneimittel binden können, sollten Patienten TRICOR mindestens 1 Stunde vor oder 4 bis 6 Stunden nach einem Gallensäure-Bindungsharz einnehmen, um eine Behinderung seiner Absorption zu vermeiden.
Colchicin
Fälle von Myopathie, einschließlich Rhabdomyolyse, wurden mit Fenofibraten berichtet, die zusammen mit Colchicin verabreicht wurden, und bei der Verschreibung von Fenofibrat mit Colchicin ist Vorsicht geboten.
Schwangerschaftskategorie C
Die Sicherheit bei schwangeren Frauen wurde nicht nachgewiesen. Es gibt keine angemessenen und gut kontrollierten Studien zu Fenofibrat bei schwangeren Frauen. Fenofibrat sollte während der Schwangerschaft nur angewendet werden, wenn der potenzielle Nutzen das potenzielle Risiko für den Fötus rechtfertigt.
Bei weiblichen Ratten, denen 15 Tage vor der Paarung durch Absetzen orale Nahrungsdosen von 15, 75 und 300 mg / kg / Tag Fenofibrat verabreicht wurden, wurde die maternale Toxizität bei 0,3-facher MRHD beobachtet, basierend auf Vergleichen der Körperoberfläche. mg / m2.
Bei trächtigen Ratten, denen während des Zeitraums der Organogenese orale Nahrungsdosen von 14, 127 und 361 mg / kg / Tag ab dem 6-15. Schwangerschaftstag verabreicht wurden, wurden bei 14 mg / kg / Tag (weniger als das 1-fache der MRHD) keine nachteiligen Entwicklungsergebnisse beobachtet basierend auf Vergleichen der Körperoberfläche; mg / m2). Bei höheren Vielfachen menschlicher Dosen wurde ein Nachweis der maternalen Toxizität beobachtet.
Bei trächtigen Kaninchen, denen ab dem Schwangerschaftstag 618 während des Zeitraums der Organogenese orale Sondendosen von 15, 150 und 300 mg / kg / Tag verabreicht wurden und die abgegeben werden durften, wurden abgebrochene Würfe mit 150 mg / kg / Tag (10-fache MRHD) beobachtet. basierend auf Vergleichen der Körperoberfläche: mg / m2). Bei 15 mg / kg / Tag (bei weniger als dem 1-fachen der MRHD, basierend auf Vergleichen der Körperoberfläche; mg / m) wurden keine Entwicklungsergebnisse beobachtet2).
Bei trächtigen Ratten, denen vom 15. Schwangerschaftstag bis zum 21. Laktationstag (Absetzen) orale Nahrungsdosen von 15, 75 und 300 mg / kg / Tag verabreicht wurden, wurde die maternale Toxizität bei weniger als dem 1-fachen der empfohlenen maximalen menschlichen Dosis (MRHD) beobachtet auf Vergleiche der Körperoberfläche; mg / m2.
Erfahrung in klinischen Studien
Da klinische Studien unter sehr unterschiedlichen Bedingungen durchgeführt werden, können in den klinischen Studien eines Arzneimittels beobachtete Nebenwirkungsraten nicht direkt mit den Raten in den klinischen Studien eines anderen Arzneimittels verglichen werden und spiegeln möglicherweise nicht die in der Praxis beobachteten Raten wider.
Unerwünschte Ereignisse, die von 2% oder mehr der mit Fenofibrat (und mehr als Placebo) behandelten Patienten während der doppelblinden, placebokontrollierten Studien unabhängig von ihrer Kausalität gemeldet wurden, sind in der folgenden Tabelle 1 aufgeführt. Unerwünschte Ereignisse führten bei 5,0% der mit Fenofibrat behandelten Patienten und bei 3,0%, die mit Placebo behandelt wurden, zum Absetzen der Behandlung. Erhöhungen der Leberfunktionstests waren die häufigsten Ereignisse, die bei 1,6% der Patienten in Doppelblindstudien zum Absetzen der Fenofibrat-Behandlung führten.
Tabelle 1. Unerwünschte Reaktionen, die von 2% oder mehr der mit Fenofibrat behandelten Patienten gemeldet wurden und während der doppelblinden, placebokontrollierten Studien größer als Placebo waren
| KÖRPERSYSTEM Unerwünschte Reaktion |
Fenofibrat * | Placebo |
| (N = 439) | (N = 439) | |
| KÖRPER ALS GANZ | ||
| Bauchschmerzen | 4,6% | 4,4% |
| Rückenschmerzen | 3,4% | 2,5% |
| Kopfschmerzen | 3,2% | 2,7% |
| DIGESTIVE | ||
| Übelkeit | 2,3% | 1,9% |
| Verstopfung | 2,1% | 1,4% |
| METABOLISCHE UND ERNÄHRUNGSWESEN | ||
| Abnormale Leberfunktionstests | 7,5% ** | 1,4% |
| Erhöhte ALT | 3,0% | 1,6% |
| Erhöhte CPK | 3,0% | 1,4% |
| Erhöhte AST | 3,4% ** | 0,5% |
| VERLETZUNG | ||
| Atemstörung | 6,2% | 5,5% |
| Rhinitis | 2,3% | 1,1% |
| * Dosierung entsprechend 160 mg TRICOR . ** Deutlich anders als Placebo. |
||
Postmarketing-Erfahrung
Die folgenden Nebenwirkungen wurden während der Anwendung von Fenofibrat nach der Zulassung festgestellt: Myalgie, Rhabdomyolyse, Pankreatitis, akutes Nierenversagen, Muskelkrampf, Hepatitis, Zirrhose, Anämie, Arthralgie, Abnahme des Hämoglobins, Abnahme des Hämatokrits, Abnahme der weißen Blutkörperchen, Asthenie und stark depressive HDL-Cholesterinspiegel. Da diese Reaktionen freiwillig von einer Population ungewisser Größe gemeldet werden, ist es nicht immer möglich, ihre Häufigkeit zuverlässig abzuschätzen oder einen ursächlichen Zusammenhang mit der Arzneimittelexposition herzustellen.
Es gibt keine spezifische Behandlung für eine Überdosierung mit TRICOR. Eine allgemeine unterstützende Behandlung des Patienten ist angezeigt, einschließlich der Überwachung der Vitalfunktionen und der Beobachtung des klinischen Status, falls eine Überdosierung auftreten sollte. Falls angezeigt, sollte die Eliminierung des nicht absorbierten Arzneimittels durch Erbrechen oder Magenspülung erreicht werden. Es sollten übliche Vorsichtsmaßnahmen getroffen werden, um die Atemwege aufrechtzuerhalten. Da Fenofibrinsäure stark an Plasmaproteine gebunden ist, sollte eine Hämodialyse nicht in Betracht gezogen werden.
Eine Vielzahl klinischer Studien hat gezeigt, dass erhöhte Spiegel von Gesamt-C, LDL-C und Apo B, einem LDL-Membrankomplex, mit menschlicher Atherosklerose verbunden sind. In ähnlicher Weise sind verringerte HDL-C-Spiegel und sein Transportkomplex Apolipoprotein A (Apo AI und Apo AII) verbunden mit der Entwicklung der Atherosklerose. Epidemiologische Untersuchungen haben ergeben, dass kardiovaskuläre Morbidität und Mortalität direkt mit dem Gesamt-C, LDL-C und TG und umgekehrt mit dem HDL-C-Spiegel variieren. Der unabhängige Effekt der Erhöhung von HDL-C oder der Senkung von Triglyceriden (TG) auf das Risiko einer kardiovaskulären Morbidität und Mortalität wurde nicht bestimmt.
Fenofibric Säure, der aktive Metabolit von Fenofibrat, führt bei behandelten Patienten zu einer Verringerung des Gesamtcholesterins, des LDL-Cholesterins, des Apolipoproteins B, der Gesamttriglyceride und des triglyceridreichen Lipoproteins (VLDL). Darüber hinaus führt die Behandlung mit Fenofibrat zu einem Anstieg des Lipoproteins (HDL) und der Apolipoproteine ApoAI und ApoAII mit hoher Dichte
Fenofibrate is a pro-drug of the active chemical moiety fenofibric acid. Fenofibrate is converted by ester hydrolysis in the body to fenofibric acid which is the active constituent measurable in the circulation.
Absorption
The absolute bioavailability of fenofibrate cannot be determined as the compound is virtually insoluble in aqueous media suitable for injection. However, fenofibrate is well absorbed from the gastrointestinal tract. Following oral administration in healthy volunteers, approximately 60% of a single dose of radiolabelled fenofibrate appeared in urine, primarily as fenofibric acid and its glucuronate conjugate, and 25% was excreted in the feces. Peak plasma levels of fenofibric acid occur within 6 to 8 hours after administration.
The absorption of fenofibrate is increased when administered with food. With fenofibrate tablets, the extent of absorption is increased by approximately 35% under fed as compared to fasting conditions.
Distribution
Upon multiple dosing of fenofibrate, fenofibric acid steady state is achieved within 5 days. Plasma concentrations of fenofibric acid at steady state are approximately double of those following a single dose. Serum protein binding was approximately 99% in normal and hyperlipidemic subjects.
Metabolism
Following oral administration, fenofibrate is rapidly hydrolyzed by esterases to the active metabolite, fenofibric acid; no unchanged fenofibrate is detected in plasma.
Fenofibric acid is primarily conjugated with glucuronic acid and then excreted in urine. A small amount of fenofibric acid is reduced at the carbonyl moiety to a benzhydrol metabolite which is, in turn, conjugated with glucuronic acid and excreted in urine.
In vivo metabolism data indicate that neither fenofibrate nor fenofibric acid undergo oxidative metabolism (e.g., cytochrome P450) to a significant extent.
Elimination
After absorption, fenofibrate is mainly excreted in the urine in the form of metabolites, primarily fenofibric acid and fenofibric acid glucuronide. After administration of radiolabelled fenofibrate, approximately 60% of the dose appeared in the urine and 25% was excreted in the feces.
Fenofibric acid is eliminated with a half-life of 20 hours, allowing once daily dosing.
However, we will provide data for each active ingredient





