


Componentes:
Método de ação:
Opção de tratamento:
Medicamente revisado por Oliinyk Elizabeth Ivanovna, Farmácia Última atualização em 10.04.2022

Atenção! As informações na página são apenas para profissionais de saúde! As informações são coletadas em fontes abertas e podem conter erros significativos! Tenha cuidado e verifique novamente todas as informações desta página!
20 principais medicamentos com os mesmos componentes:
Formas e forças de dosagem
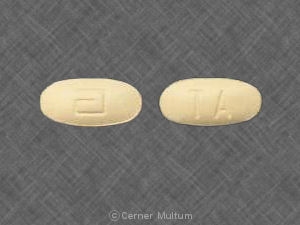
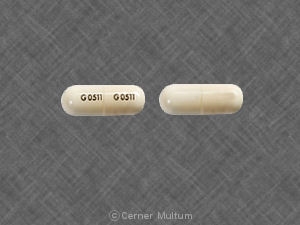
- Comprimidos amarelos de 54 mg, impressos com o logotipo "a" e as letras de identificação do código "TA".
- Comprimidos brancos de 160 mg, impressos com o logotipo "a" e as letras de identificação do código "TC".
Armazenamento e manuseio
TRICOR® (comprimidos de fenofibrato) está disponível em dois pontos fortes :
54 mg comprimidos amarelos, impressos com o logotipo “a” e as letras de identificação de código "TA", disponíveis em frascos de 90 (NDC 0074-4009-90).
160 mg comprimidos brancos, impressos com o logotipo “a” e as letras de identificação de código "TC", disponíveis em frascos de 90 (NDC 0074-4013-90).
Armazenamento
Armazenar a 25 ° C (77 ° F); excursões permitidas a 15-30 ° C (59-86 ° F).
Mantenha fora do alcance das crianças. Proteger da umidade.
Fabricado por: Laboratoires Fournier, S.A., 21300 Chenôve, França. Revisado: janeiro de 2017.
Hipercolesterolemia primária ou dislipidemia mista
TRICOR é indicado como terapia adjuvante à dieta para reduzir o colesterol lipoproteína de baixa densidade (LDL-C), colesterol total (Total-C), triglicerídeos e apolipoproteína B (Apo B) e aumentar o colesterol de lipoproteína de alta densidade (HDL-C) em pacientes adultos com hipercolesterolemia primária ou dislipidemia mista.
Hipertrigliceridemia grave
TRICOR também é indicado como terapia adjuvante à dieta para tratamento de pacientes adultos com hipertrigliceridemia grave. Melhorar o controle glicêmico em pacientes diabéticos mostrando que a quilomicronemia em jejum geralmente evita a necessidade de intervenção farmacológica.
Níveis marcadamente elevados de triglicerídeos séricos (por exemplo,. > 2.000 mg / dL) pode aumentar o risco de desenvolver pancreatite. O efeito da terapia com fenofibrato na redução desse risco não foi adequadamente estudado.
Limitações importantes de uso
O fenofibrato em uma dose equivalente a 160 mg de TRICOR não demonstrou reduzir a morbimortalidade da doença cardíaca coronária em um grande estudo controlado randomizado de pacientes com diabetes mellitus tipo 2.
Considerações gerais
Os pacientes devem ser colocados em uma dieta hipolipemiante apropriada antes de receber TRICOR e devem continuar essa dieta durante o tratamento com TRICOR. Os comprimidos TRICOR devem ser administrados com as refeições, otimizando a biodisponibilidade do medicamento.
O tratamento inicial para dislipidemia é uma terapia alimentar específica para o tipo de anormalidade lipoproteína. O excesso de peso corporal e o excesso de ingestão alcoólica podem ser fatores importantes na hipertrigliceridemia e devem ser abordados antes de qualquer terapia medicamentosa. O exercício físico pode ser uma medida auxiliar importante. Doenças contributivas à hiperlipidemia, como hipotireoidismo ou diabetes mellitus, devem ser procuradas e tratadas adequadamente. A terapia com estrogênio, diuréticos tiazídicos e betabloqueadores, às vezes estão associados a aumentos maciços nos triglicerídeos plasmáticos, especialmente em indivíduos com hipertrigliceridemia familiar. Nesses casos, a descontinuação do agente etiológico específico pode evitar a necessidade de terapia medicamentosa específica da hipertrigliceridemia.
Os níveis lipídicos devem ser monitorados periodicamente e deve-se considerar a redução da dose de TRICOR se os níveis lipídicos caírem significativamente abaixo da faixa alvo.
A terapia deve ser retirada em pacientes que não apresentam uma resposta adequada após dois meses de tratamento com a dose máxima recomendada de 160 mg uma vez ao dia.
Hipercolesterolemia primária ou dislipidemia mista
A dose inicial de TRICOR é de 160 mg uma vez ao dia.
Hipertrigliceridemia grave
A dose inicial é de 54 a 160 mg por dia. A dosagem deve ser individualizada de acordo com a resposta do paciente e deve ser ajustada, se necessário, após determinações lipídicas repetidas em intervalos de 4 a 8 semanas. A dose máxima é de 160 mg uma vez ao dia.
Função renal prejudicada
O tratamento com TRICOR deve ser iniciado na dose de 54 mg por dia em pacientes com função renal leve a moderada e aumentado somente após avaliação dos efeitos na função renal e nos níveis lipídicos nessa dose. O uso de TRICOR deve ser evitado em pacientes com insuficiência renal grave.
Pacientes geriátricos
A seleção da dose para idosos deve ser feita com base na função renal.
TRICOR está contra-indicado em :
- pacientes com insuficiência renal grave, incluindo aqueles que recebem diálise.
- pacientes com doença hepática ativa, incluindo aqueles com cirrose biliar primária e anormalidades inexplicáveis da função hepática persistente.
- pacientes com doença da vesícula biliar preexistente.
- nutrizes
- doentes com hipersensibilidade conhecida ao fenofibrato ou ácido fenofíbrico.
AVISO
Incluído como parte do "PRECAUÇÕES" Seção
PRECAUÇÕES
Mortalidade e morbidade coronariana por doença cardíaca
O efeito do TRICOR na morbimortalidade e mortalidade de doenças cardíacas coronárias e na mortalidade não cardiovascular não foi estabelecido.
O estudo Ação para controlar o risco cardiovascular no diabetes lipídico (ACCORD Lipid) foi um estudo randomizado controlado por placebo de 5518 pacientes com diabetes mellitus tipo 2 em terapia com estatina de fundo tratada com fenofibrato. A duração média do acompanhamento foi de 4,7 anos. A terapia combinada com fenofibrato e estatina mostrou uma redução de risco relativo não significativa de 8% no resultado primário dos principais eventos cardiovasculares adversos (MACE), um composto de infarto do miocárdio não fatal, acidente vascular cerebral não fatal e morte de doenças cardiovasculares (taxa de risco [HR ] 0,92, IC 95% 0,79-1,08) (p =. Em uma análise de subgrupo de gênero, a taxa de risco para o MACE em homens que receberam terapia combinada versus estatina em monoterapia foi de 0,82 (IC 95% 0,69-0,99), e a taxa de risco para o MACE em mulheres que receberam terapia combinada versus estatina em monoterapia foi de 1,38 (IC 95% 0,98-1) ). O significado clínico desse achado de subgrupo não é claro.
O estudo de Intervenção e Redução de Eventos em Diabetes (FIELD) de Fenofibrato foi um estudo randomizado, controlado por placebo, de 5 anos, de 9795 pacientes com diabetes mellitus tipo 2 tratados com fenofibrato. O fenofibrato demonstrou uma redução relativa não significativa de 11% no resultado primário de eventos de doenças cardíacas coronárias (taxa de risco [HR] 0,89, IC 95% 0,75-1,05, p = 0,16) e uma redução significativa de 11% no resultado secundário de eventos totais de doenças cardiovasculares (HR 0,89 [0,80,80,80,0,80,0,0. Houve um aumento não significativo de 11% (HR 1,11 [0,95, 1,29], p = 0,18) e 19% (HR 1,19 [0,90, 1,57], p = 0,22) na mortalidade total e coronária de doenças cardíacas, respectivamente, com fenofibrato em comparação com placebo.
Devido às semelhanças químicas, farmacológicas e clínicas entre TRICOR (comprimidos de fenofibrato), clofibrato e gemfibrozil, os achados adversos em 4 grandes estudos clínicos randomizados e controlados por placebo com esses outros medicamentos fibratos também podem ser aplicados ao TRICOR
No Projeto Coronário de Medicamentos, um grande estudo do infarto pós-miocárdio de pacientes tratados por 5 anos com clofibrato, não houve diferença na mortalidade observada entre o grupo clofibrato e o grupo placebo. Houve, no entanto, uma diferença na taxa de colelitíase e colecistite que requer cirurgia entre os dois grupos (3,0% vs. 1,8%).
Em um estudo realizado pela Organização Mundial da Saúde (OMS), 5000 indivíduos sem doença arterial coronariana conhecida foram tratados com placebo ou clofibrato por 5 anos e seguidos por mais um ano. Houve uma mortalidade por todas as causas ajustada estatisticamente significante e maior idade no grupo clofibrato em comparação com o grupo placebo (5,70% vs. 3,96%, p = <0,01). O excesso de mortalidade ocorreu devido a um aumento de 33% nas causas não cardiovasculares, incluindo malignidade, complicações pós-colecistectomia e pancreatite. Isso pareceu confirmar o maior risco de doença da vesícula biliar observado em pacientes tratados com clofibrato estudados no Projeto Coronary Drug.
O Estudo do Coração de Helsinque foi um grande estudo (n = 4081) de homens de meia idade sem histórico de doença arterial coronariana. Os indivíduos receberam placebo ou gemfibrozil por 5 anos, com uma extensão aberta de 3,5 anos depois. A mortalidade total foi numericamente maior no grupo de randomização do gemfibrozil, mas não obteve significância estatística (p = 0,19, intervalo de confiança de 95% para o risco relativo G: P = 0,91-1,64). Embora as mortes por câncer tenham aumentado no grupo gemfibrozil (p = 0,11), os cânceres (excluindo carcinoma basocelular) foram diagnosticados com a mesma frequência nos dois grupos de estudo. Devido ao tamanho limitado do estudo, o risco relativo de morte por qualquer causa não mostrou ser diferente do observado nos dados de acompanhamento de 9 anos do estudo da Organização Mundial da Saúde (RR = 1,29).
Um componente secundário de prevenção do Estudo do Coração de Helsinque registrou homens de meia idade excluídos do estudo de prevenção primária devido a doença cardíaca coronária conhecida ou suspeita. Os indivíduos receberam gemfibrozil ou placebo por 5 anos. Embora as mortes cardíacas tenham tendido mais alto no grupo gemfibrozil, isso não foi estatisticamente significativo (taxa de risco 2,2, intervalo de confiança de 95%: 0,94-5,05). A taxa de cirurgia da vesícula biliar não foi estatisticamente significativa entre os grupos de estudo, mas teve uma tendência maior no grupo gemfibrozil (1,9% vs. 0,3%, p = 0,07).
Músculo esquelético
Os fibratos aumentam o risco de miopatia e têm sido associados à rabdomiólise. O risco de toxicidade muscular grave parece aumentar em pacientes idosos e em pacientes com diabetes, insuficiência renal ou hipotireoidismo.
A miopatia deve ser considerada em qualquer paciente com mialgias difusas, sensibilidade ou fraqueza muscular e / ou elevações acentuadas dos níveis de creatina fosfoquinase (CPK).
Os pacientes devem ser aconselhados a relatar prontamente dores musculares inexplicáveis, sensibilidade ou fraqueza, principalmente se acompanhados de mal-estar ou febre. Os níveis de CPK devem ser avaliados em pacientes que relatam esses sintomas, e a terapia com TRICOR deve ser descontinuada se ocorrerem níveis acentuadamente elevados de CPK ou se houver suspeita ou diagnóstico de miopatia / miosite.
Dados de estudos observacionais indicam que o risco de rabdomiólise aumenta quando os fibratos, em particular o gemfibrozil, são co-administrados com um inibidor da HMG-CoA redutase (estatina). A combinação deve ser evitada, a menos que o benefício de outras alterações nos níveis lipídicos provavelmente supere o risco aumentado dessa combinação de medicamentos.
Casos de miopatia, incluindo rabdomiólise, foram relatados com fenofibratos co-administrados com colchicina, e deve-se ter cautela ao prescrever fenofibrato com colchicina.
Função hepática
O fenofibrato em doses equivalentes a 107 mg a 160 mg de TRICOR por dia foi associado a aumentos nas transaminases séricas [AST (SGOT) ou ALT (SGPT)]. Em uma análise conjunta de 10 estudos controlados por placebo, ocorreram aumentos para> 3 vezes o limite superior do normal em 5,3% dos pacientes que tomaram fenofibrato versus 1,1% dos pacientes tratados com placebo.
Quando as determinações da transaminase foram seguidas após a descontinuação do tratamento ou durante o tratamento continuado, geralmente era observado um retorno aos limites normais. A incidência de aumentos nas transaminases relacionadas à terapia com fenofibrato parece estar relacionada à dose. Em um estudo de 8 semanas sobre a dose, a incidência de elevações de ALT ou AST para pelo menos três vezes o limite superior do normal foi de 13% em pacientes que receberam doses equivalentes a 107 mg a 160 mg de TRICOR por dia e foi de 0% naqueles que receberam doses equivalentes a 54 mg ou menos de TRICOR por dia, ou placebo. Hepatite hepatocelular, crônica ativa e colestática associada à terapia com fenofibrato foram relatadas após exposições de semanas a vários anos. Em casos extremamente raros, foi relatada cirrose em associação com hepatite ativa crônica.
O monitoramento periódico básico e regular da função hepática, incluindo a ALT sérica (SGPT), deve ser realizado durante o período de terapia com TRICOR, e a terapia é interrompida se os níveis enzimáticos persistirem acima de três vezes o limite normal.
Creatinina sérica
Elevações na creatinina sérica foram relatadas em pacientes em uso de fenofibrato. Essas elevações tendem a retornar à linha de base após a descontinuação do fenofibrato. O significado clínico dessas observações é desconhecido. Monitore a função renal em pacientes com insuficiência renal em uso de TRICOR. O monitoramento renal também deve ser considerado em pacientes em uso de TRICOR em risco de insuficiência renal, como idosos e pacientes com diabetes.
Colelitíase
O fenofibrato, como o clofibrato e o gemfibrozil, pode aumentar a excreção de colesterol na bílis, levando à colelitíase. Se houver suspeita de colelitíase, são indicados estudos de vesícula biliar. A terapia com TRICOR deve ser descontinuada se forem encontradas cálculos biliares.
Anticoagulantes de cumarina
Deve-se ter cuidado quando os anticoagulantes da cumarina são administrados em conjunto com o TRICOR devido à potencialização dos efeitos anticoagulantes do tipo cumarina no prolongamento do Tempo de Protrombina / Índice Normalizado Internacional (PT / INR). Para evitar complicações hemorrágicas, recomenda-se o monitoramento frequente de PT / INR e o ajuste da dose do anticoagulante até que o PT / INR se estabilize.
Pancreatite
Foi relatada pancreatite em pacientes em uso de fenofibrato, gemfibrozil e clofibrato. Essa ocorrência pode representar uma falha de eficácia em pacientes com hipertrigliceridemia grave, um efeito direto do medicamento ou um fenômeno secundário mediado pela formação de pedra do trato biliar ou lodo com obstrução do ducto biliar comum.
Alterações hematológicas
Foram observadas reduções leves a moderadas de hemoglobina, hematócrito e glóbulos brancos em pacientes após o início da terapia com fenofibrato. No entanto, esses níveis se estabilizam durante a administração a longo prazo. Trombocitopenia e agranulocitose foram relatadas em indivíduos tratados com fenofibrato. O monitoramento periódico das contagens de glóbulos vermelhos e brancos é recomendado durante os primeiros 12 meses de administração do TRICOR.
Reações de hipersensibilidade
Foram relatadas reações agudas de hipersensibilidade, como a síndrome de Stevens-Johnson e necrólise epidérmica tóxica que requerem hospitalização do paciente e tratamento com esteróides em indivíduos tratados com fenofibratos. Urticária foi vista em 1.1 vs. 0% e erupção cutânea em 1,4 vs. 0,8% dos pacientes com fenofibrato e placebo, respectivamente, em ensaios controlados.
Doença venotromboembólica
No estudo FIELD, embolia pulmonar (PE) e trombose venosa profunda (TVP) foram observadas em taxas mais altas no fenofibrato do que no grupo tratado com placebo. Dos 9.795 pacientes inscritos no FIELD, havia 4.900 no grupo placebo e 4.895 no grupo fenofibrato. Para DVT, houve 48 eventos (1%) no grupo placebo e 67 (1%) no grupo fenofibrato (p = 0,074); e para PE, houve 32 (0,7%) eventos no grupo placebo e 53 (1%) no grupo fenofibrato (p = 0,022).
No Projeto Coronário de Medicamentos, uma proporção maior do grupo clofibrato apresentou embolia ou tromboflebite pulmonar definitiva ou suspeita fatal ou não fatal do que o grupo placebo (5,2% vs. 3,3% em cinco anos; p <0,01).
Diminuições paradoxais nos níveis de colesterol HDL
Houve relatos pós-comercialização e ensaios clínicos de reduções graves nos níveis de colesterol HDL (tão baixo quanto 2 mg / dL) ocorrendo em pacientes diabéticos e não diabéticos iniciados em terapia com fibratos. A diminuição do HDL-C é refletida por uma diminuição na apolipoproteína A1. Foi relatado que essa diminuição ocorre dentro de 2 semanas a anos após o início da terapia com fibrato. Os níveis de HDL-C permanecem deprimidos até a terapia com fibratos ser retirada; a resposta à retirada da terapia com fibrato é rápida e sustentada. O significado clínico dessa diminuição no HDL-C é desconhecido. Recomenda-se que os níveis de HDL-C sejam verificados nos primeiros meses após o início da terapia com fibratos. Se um nível de HDL-C gravemente deprimido for detectado, a terapia com fibrato deve ser retirada e o nível de HDL-C monitorado até retornar à linha de base, e a terapia com fibrato não deve ser reiniciada.
Toxicologia Não Clínica
Carcinogênese e mutagênese e comprometimento da fertilidade
Dois estudos de carcinogenicidade dietética foram realizados em ratos com fenofibrato. No primeiro estudo de 24 meses, os ratos Wistar foram doseados com fenofibrato a 10, 45 e 200 mg / kg / dia, aproximadamente 0,3, 1 e 6 vezes a dose máxima recomendada humana (MRHD), com base nas comparações da área da superfície corporal (mg / m2). Na dose de 200 mg / kg / dia (6 vezes o MRHD), a incidência de carcinomas no fígado aumentou significativamente em ambos os sexos. Um aumento estatisticamente significativo nos carcinomas pancreáticos foi observado em homens com 1 e 6 vezes o MRHD; foi observado um aumento nos adenomas pancreáticos e nos tumores benignos de células intersticiais testiculares em 6 vezes o MRHD nos homens. Em um segundo estudo de carcinogenicidade em ratos de 24 meses em uma cepa diferente de ratos (Sprague-Dawley) doses de 10 e 60 mg / kg / dia (0,3 e 2 vezes o MRHD) produziu aumentos significativos na incidência de adenomas acinários pancreáticos em ambos os sexos e aumentos nos tumores de células intersticiais testiculares em homens em 2 vezes o MRHD
Foi realizado um estudo de carcinogenicidade de 117 semanas em ratos comparando três medicamentos: fenofibrato 10 e 60 mg / kg / dia (0,3 e 2 vezes o MRHD), clofibrato (400 mg / kg / dia; 2 vezes a dose humana) e gemfibrozil (250 mg / kg / dia; 2 vezes a dose humana, com base em mg2 área de superfície). O fenofibrato aumentou os adenomas acinares pancreáticos em ambos os sexos. O clofibrato aumentou o carcinoma hepatocelular e adenomas acinários pancreáticos em homens e nódulos neoplásicos hepáticos em mulheres. O gemfibrozil aumentou os nódulos neoplásicos hepáticos em homens e mulheres, enquanto os três medicamentos aumentaram os tumores das células intersticiais testiculares em homens.
Em um estudo de 21 meses em camundongos CF-1, fenofibrato 10, 45 e 200 mg / kg / dia (aproximadamente 0,2, 1 e 3 vezes o MRHD com base em mg / m2 área de superfície) aumentou significativamente os carcinomas hepáticos em ambos os sexos em 3 vezes o MRHD. Em um segundo estudo de 18 meses, com 10, 60 e 200 mg / kg / dia, o fenofibrato aumentou significativamente os carcinomas hepáticos em camundongos machos e adenomas hepáticos em camundongos fêmeas 3 vezes o MRHD
Estudos de microscopia eletrônica demonstraram proliferação peroxissômica após administração de fenofibrato no rato. Um estudo adequado para testar a proliferação de peroxissomos em humanos não foi realizado, mas alterações na morfologia e nos números de peroxissomos foram observadas em humanos após o tratamento com outros membros da classe de fibratos quando as biópsias hepáticas foram comparadas antes e após o tratamento no mesmo indivíduo.
Mutagênese
Demonstrou-se que o fenofibrato é desprovido de potencial mutagênico nos seguintes testes: Ames, linfoma de camundongo, aberração cromossômica e síntese de DNA não programada em hepatócitos primários de ratos.
Compromisso de fertilidade
Em estudos de fertilidade, os ratos receberam doses alimentares orais de fenofibrato, os machos receberam 61 dias antes do acasalamento e as fêmeas 15 dias antes do acasalamento através do desmame, o que não resultou em efeito adverso na fertilidade em doses de até 300 mg / kg / dia (~ 10 vezes o MRHD, com base em mg / m2 comparações de área de superfície).
Use em populações específicas
Gravidez
Categoria de gravidez C
A segurança em mulheres grávidas não foi estabelecida. Não há estudos adequados e bem controlados de fenofibrato em mulheres grávidas. O fenofibrato deve ser usado durante a gravidez apenas se o benefício potencial justificar o risco potencial para o feto.
Em ratos fêmeas que receberam doses alimentares orais de 15, 75 e 300 mg / kg / dia de fenofibrato de 15 dias antes do acasalamento através do desmame, foi observada toxicidade materna em 0,3 vezes o MRHD, com base nas comparações da área da superfície corporal; mg / m2.
Em ratos grávidas que receberam doses alimentares orais de 14, 127 e 361 mg / kg / dia do dia da gestação 6-15 durante o período de organogênese, não foram observados achados adversos no desenvolvimento em 14 mg / kg / dia (menos de 1 vezes o MRHD, com base nas comparações da área da superfície corporal; mg / m2). Em múltiplos mais altos de doses humanas, foram observadas evidências de toxicidade materna.
Em coelhos prenhes que receberam doses de gavagem oral de 15, 150 e 300 mg / kg / dia a partir do dia da gestação 618 durante o período de organogênese e permitiram o parto, foram observadas ninhadas abortadas a 150 mg / kg / dia (10 vezes o MRHD, com base nas comparações da área da superfície corporal: mg / m2). Não foram observados achados de desenvolvimento em 15 mg / kg / dia (em menos de 1 vezes o MRHD, com base em comparações de área da superfície corporal; mg / m2).
Em ratos grávidas que receberam doses alimentares orais de 15, 75 e 300 mg / kg / dia desde o dia da gestação 15 até o dia da lactação 21 (desmame), a toxicidade materna foi observada em menos de 1 vezes a dose humana máxima recomendada (MRHD), com base nas comparações da área da superfície corporal; mg / m2.
Mães de enfermagem
O fenofibrato não deve ser usado em nutrizes. Deve-se tomar uma decisão sobre interromper a amamentação ou interromper o medicamento, levando em consideração a importância do medicamento para a mãe.
Uso pediátrico
Segurança e eficácia não foram estabelecidas em pacientes pediátricos.
Uso geriátrico
Sabe-se que o ácido fenofíbrico é substancialmente excretado pelo rim, e o risco de reações adversas a este medicamento pode ser maior em pacientes com insuficiência renal. A exposição ao ácido fenofíbrico não é influenciada pela idade. Como pacientes idosos têm maior incidência de insuficiência renal, a seleção da dose para idosos deve ser feita com base na função renal. Pacientes idosos com função renal normal não devem exigir modificações na dose. Considere monitorar a função renal em pacientes idosos que tomam TRICOR
Compromisso renal
O uso de TRICOR deve ser evitado em pacientes com insuficiência renal grave. É necessária redução da dose em pacientes com insuficiência renal leve a moderada. Recomenda-se o monitoramento da função renal em pacientes com insuficiência renal.
Compromisso hepático
O uso de TRICOR não foi avaliado em indivíduos com insuficiência hepática.
EFEITOS SECUNDÁRIOS
Experiência em ensaios clínicos
Como os estudos clínicos são realizados em condições muito variadas, as taxas de reação adversa observadas nos estudos clínicos de um medicamento não podem ser diretamente comparadas às taxas nos estudos clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
Eventos adversos relatados por 2% ou mais dos pacientes tratados com fenofibrato (e maior que o placebo) durante os ensaios duplo-cegos, controlados por placebo, independentemente da causalidade, estão listados na Tabela 1 abaixo. Eventos adversos levaram à descontinuação do tratamento em 5,0% dos pacientes tratados com fenofibrato e em 3,0% tratados com placebo. Aumentos nos testes de função hepática foram os eventos mais frequentes, causando a descontinuação do tratamento com fenofibrato em 1,6% dos pacientes em ensaios duplo-cegos.
Quadro 1. Reações adversas relatadas por 2% ou mais dos pacientes tratados com fenofibrato e maior que o placebo durante os ensaios duplo-cegos e controlados por placebo
| SISTEMA DE CORPO Reação Adversa |
Fenofibrato * | Placebo |
| (N = 439) | (N = 439) | |
| CORPO COMO UM TODO | ||
| Dor abdominal | 4,6% | 4,4% |
| Dor nas costas | 3,4% | 2,5% |
| Dor de cabeça | 3,2% | 2,7% |
| DIGESTIVO | ||
| Náusea | 2,3% | 1,9% |
| Constipação | 2,1% | 1,4% |
| TRANSTORNOS METABÓLICOS E NUTRIÇÃO | ||
| Testes anormais da função hepática | 7,5% ** | 1,4% |
| ALT aumentado | 3,0% | 1,6% |
| CPK aumentado | 3,0% | 1,4% |
| Aumento do AST | 3,4% ** | 0,5% |
| RESPIRATÓRIO | ||
| Transtorno Respiratório | 6,2% | 5,5% |
| Rinite | 2,3% | 1,1% |
| * Dosagem equivalente a 160 mg TRICOR . ** Significativamente diferente do Placebo. |
||
Experiência pós-comercialização
As seguintes reações adversas foram identificadas durante o uso pós-aprovação de fenofibrato: mialgia, rabdomiólise, pancreatite, insuficiência renal aguda, espasmo muscular, hepatite, cirrose, anemia, artralgia, diminuição da hemoglobina, diminuição do hematócrito, diminuição dos glóbulos brancos, astenia e níveis severamente deprimidos de HDL-colesterol. Como essas reações são relatadas voluntariamente a partir de uma população de tamanho incerto, nem sempre é possível estimar com segurança sua frequência ou estabelecer uma relação causal com a exposição a medicamentos.
INTERAÇÕES DE DROGAS
Anticoagulantes de cumarina
A potencialização dos efeitos anticoagulantes do tipo cumarina foi observada com o prolongamento do PT / INR
Deve-se ter cuidado quando anticoagulantes de cumarina são administrados em conjunto com TRICOR. A dosagem dos anticoagulantes deve ser reduzida para manter o PT / INR no nível desejado para evitar complicações hemorrágicas. As determinações freqüentes de PT / INR são aconselháveis até que seja definitivamente determinado que o PT / INR se estabilizou.
Imunossupressores
Imunossupressores como ciclosporina e tacrolimus podem produzir nefrotoxicidade com reduções na depuração da creatinina e aumentos na creatinina sérica, e como a excreção renal é a principal via de eliminação de medicamentos fibratos, incluindo TRICOR, existe o risco de uma interação levar à deterioração da função renal. Os benefícios e riscos do uso de TRICOR (comprimidos de fenofibrato) com imunossupressores e outros agentes potencialmente nefrotóxicos devem ser cuidadosamente considerados, e a menor dose efetiva empregada e a função renal monitorada.
Resinas de ligação a ácidos biliares
Como as resinas de ligação ao ácido biliar podem ligar outros medicamentos administrados simultaneamente, os pacientes devem tomar TRICOR pelo menos 1 hora antes ou 4 a 6 horas após uma resina de ligação ao ácido biliar para evitar impedir sua absorção.
Colchicina
Casos de miopatia, incluindo rabdomiólise, foram relatados com fenofibratos co-administrados com colchicina, e deve-se ter cautela ao prescrever fenofibrato com colchicina.
Categoria de gravidez C
A segurança em mulheres grávidas não foi estabelecida. Não há estudos adequados e bem controlados de fenofibrato em mulheres grávidas. O fenofibrato deve ser usado durante a gravidez apenas se o benefício potencial justificar o risco potencial para o feto.
Em ratos fêmeas que receberam doses alimentares orais de 15, 75 e 300 mg / kg / dia de fenofibrato de 15 dias antes do acasalamento através do desmame, foi observada toxicidade materna em 0,3 vezes o MRHD, com base nas comparações da área da superfície corporal; mg / m2.
Em ratos grávidas que receberam doses alimentares orais de 14, 127 e 361 mg / kg / dia do dia da gestação 6-15 durante o período de organogênese, não foram observados achados adversos no desenvolvimento em 14 mg / kg / dia (menos de 1 vezes o MRHD, com base nas comparações da área da superfície corporal; mg / m2). Em múltiplos mais altos de doses humanas, foram observadas evidências de toxicidade materna.
Em coelhos prenhes que receberam doses de gavagem oral de 15, 150 e 300 mg / kg / dia a partir do dia da gestação 618 durante o período de organogênese e permitiram o parto, foram observadas ninhadas abortadas a 150 mg / kg / dia (10 vezes o MRHD, com base nas comparações da área da superfície corporal: mg / m2). Não foram observados achados de desenvolvimento em 15 mg / kg / dia (em menos de 1 vezes o MRHD, com base em comparações de área da superfície corporal; mg / m2).
Em ratos grávidas que receberam doses alimentares orais de 15, 75 e 300 mg / kg / dia desde o dia da gestação 15 até o dia da lactação 21 (desmame), a toxicidade materna foi observada em menos de 1 vezes a dose humana máxima recomendada (MRHD), com base nas comparações da área da superfície corporal; mg / m2.
Experiência em ensaios clínicos
Como os estudos clínicos são realizados em condições muito variadas, as taxas de reação adversa observadas nos estudos clínicos de um medicamento não podem ser diretamente comparadas às taxas nos estudos clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
Eventos adversos relatados por 2% ou mais dos pacientes tratados com fenofibrato (e maior que o placebo) durante os ensaios duplo-cegos, controlados por placebo, independentemente da causalidade, estão listados na Tabela 1 abaixo. Eventos adversos levaram à descontinuação do tratamento em 5,0% dos pacientes tratados com fenofibrato e em 3,0% tratados com placebo. Aumentos nos testes de função hepática foram os eventos mais frequentes, causando a descontinuação do tratamento com fenofibrato em 1,6% dos pacientes em ensaios duplo-cegos.
Quadro 1. Reações adversas relatadas por 2% ou mais dos pacientes tratados com fenofibrato e maior que o placebo durante os ensaios duplo-cegos e controlados por placebo
| SISTEMA DE CORPO Reação Adversa |
Fenofibrato * | Placebo |
| (N = 439) | (N = 439) | |
| CORPO COMO UM TODO | ||
| Dor abdominal | 4,6% | 4,4% |
| Dor nas costas | 3,4% | 2,5% |
| Dor de cabeça | 3,2% | 2,7% |
| DIGESTIVO | ||
| Náusea | 2,3% | 1,9% |
| Constipação | 2,1% | 1,4% |
| TRANSTORNOS METABÓLICOS E NUTRIÇÃO | ||
| Testes anormais da função hepática | 7,5% ** | 1,4% |
| ALT aumentado | 3,0% | 1,6% |
| CPK aumentado | 3,0% | 1,4% |
| Aumento do AST | 3,4% ** | 0,5% |
| RESPIRATÓRIO | ||
| Transtorno Respiratório | 6,2% | 5,5% |
| Rinite | 2,3% | 1,1% |
| * Dosagem equivalente a 160 mg TRICOR . ** Significativamente diferente do Placebo. |
||
Experiência pós-comercialização
As seguintes reações adversas foram identificadas durante o uso pós-aprovação de fenofibrato: mialgia, rabdomiólise, pancreatite, insuficiência renal aguda, espasmo muscular, hepatite, cirrose, anemia, artralgia, diminuição da hemoglobina, diminuição do hematócrito, diminuição dos glóbulos brancos, astenia e níveis severamente deprimidos de HDL-colesterol. Como essas reações são relatadas voluntariamente a partir de uma população de tamanho incerto, nem sempre é possível estimar com segurança sua frequência ou estabelecer uma relação causal com a exposição a medicamentos.
Não há tratamento específico para sobredosagem com TRICOR. O atendimento geral de suporte ao paciente é indicado, incluindo monitoramento de sinais vitais e observação do estado clínico, caso ocorra uma overdose. Se indicado, a eliminação do medicamento não absorvido deve ser alcançada por emese ou lavagem gástrica; precauções usuais devem ser observadas para manter as vias aéreas. Como o ácido fenofíbrico está altamente ligado às proteínas plasmáticas, a hemodiálise não deve ser considerada.
Uma variedade de estudos clínicos demonstrou que níveis elevados de C total, LDL-C e apo B, um complexo de membrana LDL, estão associados à aterosclerose humana. Da mesma forma, níveis reduzidos de HDL-C e seu complexo de transporte, apolipoproteína A (apo AI e apo AII) estão associados com o desenvolvimento de aterosclerose. Investigações epidemiológicas estabeleceram que a morbimortalidade cardiovascular varia diretamente com o nível total de C, LDL-C e TG e inversamente com o nível de HDL-C. O efeito independente de aumentar o HDL-C ou diminuir os triglicerídeos (TG) no risco de morbimortalidade cardiovascular não foi determinado.
O ácido fenofíbrico, o metabolito ativo do fenofibrato, produz reduções no colesterol total, colesterol LDL, apolipoproteína B, triglicerídeos totais e lipoproteína rica em triglicerídeos (VLDL) em pacientes tratados. Além disso, o tratamento com fenofibrato resulta em aumentos na lipoproteína de alta densidade (HDL) e apolipoproteínas apoAI e apoAII
O fenofibrato é um pró-medicamento do ácido fenofíbrico ativo da fração química. O fenofibrato é convertido por hidrólise do éster no corpo em ácido fenofíbrico, que é o constituinte ativo mensurável na circulação.
Absorção
A biodisponibilidade absoluta do fenofibrato não pode ser determinada, pois o composto é praticamente insolúvel em meio aquoso adequado para injeção. No entanto, o fenofibrato é bem absorvido pelo trato gastrointestinal. Após administração oral em voluntários saudáveis, aproximadamente 60% de uma dose única de fenofibrato radiomarcado apareceu na urina, principalmente como ácido fenofíbrico e seu conjugado glucuronato, e 25% foram excretados nas fezes. Os níveis plasmáticos máximos de ácido fenofíbrico ocorrem dentro de 6 a 8 horas após a administração.
A absorção do fenofibrato aumenta quando administrada com alimentos. Com os comprimidos de fenofibrato, a extensão da absorção é aumentada em aproximadamente 35% sob alimentação, em comparação com as condições de jejum.
Distribuição
Após doses múltiplas de fenofibrato, o estado estacionário do ácido fenofíbrico é alcançado em 5 dias. As concentrações plasmáticas de ácido fenofíbrico no estado estacionário são aproximadamente o dobro daquelas após uma dose única. A ligação às proteínas séricas foi de aproximadamente 99% em indivíduos normais e hiperlipidêmicos.
Metabolismo
Após administração oral, o fenofibrato é rapidamente hidrolisado por esterases no metabolito ativo, ácido fenofibrico; nenhum fenofibrato inalterado é detectado no plasma.
O ácido fenofíbrico é conjugado principalmente com ácido glucurônico e depois excretado na urina. Uma pequena quantidade de ácido fenofíbrico é reduzida na fração carbonil para um metabólito do benzidrol que é, por sua vez, conjugado com ácido glucurônico e excretado na urina.
In vivo os dados do metabolismo indicam que nem o fenofibrato nem o ácido fenofíbrico sofrem metabolismo oxidativo (por exemplo,., citocromo P450) em uma extensão significativa.
Eliminação
Após a absorção, o fenofibrato é principalmente excretado na urina na forma de metabólitos, principalmente ácido fenofíbrico e glucuronido do ácido fenofíbrico. Após a administração de fenofibrato radiomarcado, aproximadamente 60% da dose apareceu na urina e 25% foram excretados nas fezes.
O ácido fenofíbrico é eliminado com uma meia-vida de 20 horas, permitindo uma dose diária.
However, we will provide data for each active ingredient





