
コンポーネント:
治療オプション:
Militian Inessa Mesropovna 、薬局による医学的評価、 最終更新日:15.03.2022

アテンション! そのこのページの情報は医療専門家のみを対象としています! その情報が収集したオープン源を含めることが可能である重大な誤差! 注意して、このページ上のすべての情報を再確認してください!
投薬形態と強さ。
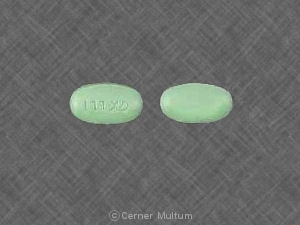
TRIZIVIR。 錠剤には、アバカビルとして300 mgのアバカビルが含まれています。 硫酸塩、150 mgのラミブジン、300 mgのジドブジン。. タブレットはです。 青緑色、カプセル型、フィルムコーティング、1つに「GX LL1」が刻印されています。 裏側にマーキングのない側。.
保管と取り扱い。
TRIZIVIR。 タブレットとして入手できます。. 各タブレットが含まれています。 アバカビル硫酸塩として300 mgのアバカビル、150 mgのラミブジン、300 mgのアバカビル。 ジドブジン。. 錠剤は青緑のカプセル型で、フィルムコーティングされています。 片側にGX LL1が刻印されており、裏側にマーキングはありません。. 彼ら。 次のようにパッケージ化されています。
60錠のボトル(。NDC。 49702-217-18)。.
25°C(77°F)で保管してください。 15°〜30°Cの遠足が許可されています。 (59°〜86°F)( USP制御室温。).
製造元:ViiV Healthcare、Research Triangle。 パーク、ノースカロライナ27709作成者:GlaxoSmithKline Research Triangle Park、ノースカロライナ27709。. 改訂:2015年9月。
TRIZIVIRは他のものと組み合わせて示されます。 ヒト免疫不全ウイルスタイプの治療のための抗レトロウイルス薬または単独。 1(HIV-1)感染。.
使用の制限。
- TRIZIVIRのみの使用に関するデータは限られています。 ベースラインのウイルス量レベルが高い患者(100,000コピーを超える患者)。 mLあたり)。.
開始前のHLA-B * 5701 Alleleのスクリーニング。 TRIZIVIR。
開始前のHLA-B * 5701対立遺伝子の画面。 TRIZIVIRによる治療
成人および小児患者に推奨される投与量。 計量少なくとも40 kg。
TRIZIVIRの推奨用量は1錠です。 食事の有無にかかわらず、1日2回経口投与。.
投与量の調整がないため、推奨されません。
TRIZIVIRは固定用量のタブレットであり、そうすることができないからです。 用量調整、TRIZIVIRは以下では推奨されません。
- 体重が40 kg未満の小児患者。
- クレアチニンクリアランスが50 mL未満の患者。 分。
- 軽度の肝機能障害のある患者。. TRIZIVIRです。 中等度または重度の肝機能障害のある患者には禁 ⁇ 。.
TRIZIVIRは患者には禁 ⁇ です:。
- HLA-B * 5701対立遺伝子を持っている人。.
- アバカビル、ラミブジン、またはジドブジンに対する以前の過敏反応。.
- 中等度または重度の肝機能障害がある。.
WARNINGS
Included as part of the PRECAUTIONS section.
PRECAUTIONS
Hypersensitivity Reactions
Serious and sometimes fatal hypersensitivity reactions have occurred with abacavir, a component of TRIZIVIR. These hypersensitivity reactions have included multi-organ failure and anaphylaxis and typically occurred within the first 6 weeks of treatment with abacavir (median time to onset was 9 days); although abacavir hypersensitivity reactions have occurred any time during treatment. Patients who carry the HLA-B*5701 allele are at a higher risk of abacavir hypersensitivity reactions; although, patients who do not carry the HLA-B*5701 allele have developed hypersensitivity reactions. Hypersensitivity to abacavir was reported in approximately 206 (8%) of 2,670 patients in 9 clinical trials with abacavir-containing products where HLA-B*5701 screening was not performed. The incidence of suspected abacavir hypersensitivity reactions in clinical trials was 1% when subjects carrying the HLA-B*5701 allele were excluded. In any patient treated with abacavir, the clinical diagnosis of hypersensitivity reaction must remain the basis of clinical decision making.
Due to the potential for severe, serious, and possibly fatal hypersensitivity reactions with abacavir:
- All patients should be screened for the HLA-B*5701 allele prior to initiating therapy with TRIZIVIR or reinitiation of therapy with TRIZIVIR, unless patients have a previously documented HLA-B*5701 allele assessment.
- TRIZIVIR is contraindicated in patients with a prior hypersensitivity reaction to abacavir and in HLA-B*5701-positive patients.
- Before starting TRIZIVIR, review medical history for prior exposure to any abacavircontaining product. NEVER restart TRIZIVIR or any other abacavir-containing product following a hypersensitivity reaction to abacavir, regardless of HLA-B*5701 status.
- To reduce the risk of a life-threatening hypersensitivity reaction, regardless of HLA-B*5701 status, discontinue TRIZIVIR immediately if a hypersensitivity reaction is suspected, even when other diagnoses are possible (e.g., acute onset respiratory diseases such as pneumonia, bronchitis, pharyngitis, or influenza; gastroenteritis; or reactions to other medications).
- If a hypersensitivity reaction cannot be ruled out, do not restart TRIZIVIR or any other abacavir-containing products because more severe symptoms, which may include life-threatening hypotension and death, can occur within hours.
- If a hypersensitivity reaction is ruled out, patients may restart TRIZIVIR. Rarely, patients who have stopped abacavir for reasons other than symptoms of hypersensitivity have also experienced life-threatening reactions within hours of reinitiating abacavir therapy. Therefore, reintroduction of TRIZIVIR or any other abacavir-containing product is recommended only if medical care can be readily accessed.
- A Medication Guide and Warning Card that provide information about recognition of abacavir hypersensitivity reactions should be dispensed with each new prescription and refill.
Hematologic Toxicity/Bone Marrow Suppression
Zidovudine, a component of TRIZIVIR, has been associated with hematologic toxicity including neutropenia and anemia, particularly in patients with advanced HIV-1 disease. TRIZIVIR should be used with caution in patients who have bone marrow compromise evidenced by granulocyte count less than 1,000 cells per mm³ or hemoglobin less than 9.5 grams per dL.
Frequent blood counts are strongly recommended in patients with advanced HIV-1 disease who are treated with TRIZIVIR. Periodic blood counts are recommended for other HIV-1-infected patients. If anemia or neutropenia develops, dosage interruption may be needed.
Myopathy
Myopathy and myositis, with pathological changes similar to that produced by HIV-1 disease, have been associated with prolonged use of zidovudine, and therefore may occur with therapy with TRIZIVIR.
Lactic Acidosis And Severe Hepatomegaly With Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues and other antiretrovirals. See full prescribing information for ZIAGEN® (abacavir), EPIVIR® (lamivudine), and RETROVIR® (zidovudine). Treatment with TRIZIVIR should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Patients With Hepatitis B Virus Co-infection
Posttreatment Exacerbations of Hepatitis
Clinical and laboratory evidence of exacerbations of hepatitis have occurred after discontinuation of lamivudine. See full prescribing information for EPIVIR (lamivudine). Patients should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment.
Emergence of Lamivudine-resistant HBV
Safety and efficacy of lamivudine have not been established for treatment of chronic hepatitis B in subjects dually infected with HIV-1 and HBV. Emergence of hepatitis B virus variants associated with resistance to lamivudine has been reported in HIV–1-infected subjects who have received lamivudine-containing antiretroviral regimens in the presence of concurrent infection with hepatitis B virus. See full prescribing information for EPIVIR (lamivudine).
Use With Interferon-And Ribavirin-Based Regimens
Patients receiving interferon alfa with or without ribavirin and TRIZIVIR should be closely monitored for treatment-associated toxicities, especially hepatic decompensation, neutropenia, and anemia. See full prescribing information for EPIVIR (lamivudine) and RETROVIR (zidovudine). Discontinuation of TRIZIVIR should be considered as medically appropriate. Dose reduction or discontinuation of interferon alfa, ribavirin, or both should also be considered if worsening clinical toxicities are observed, including hepatic decompensation (e.g., Child-Pugh greater than 6) (see full prescribing information for interferon and ribavirin).
Exacerbation of anemia has been reported in HIV-1/HCV co-infected patients receiving ribavirin and zidovudine. Coadministration of ribavirin and TRIZIVIR is not advised.
Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including TRIZIVIR. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and “cushingoid appearance” have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Myocardial Infarction
In a published prospective, observational, epidemiological trial designed to investigate the rate of myocardial infarction (MI) in patients on combination antiretroviral therapy, the use of abacavir within the previous 6 months was correlated with an increased risk of MI. In a sponsor-conducted pooled analysis of clinical trials, no excess risk of MI was observed in abacavir-treated subjects as compared with control subjects. In totality, the available data from the observational cohort and from clinical trials are inconclusive.
As a precaution, the underlying risk of coronary heart disease should be considered when prescribing antiretroviral therapies, including abacavir, and action taken to minimize all modifiable risk factors (e.g., hypertension, hyperlipidemia, diabetes mellitus, smoking).
Therapy-Experienced Patients
In clinical trials, subjects with prolonged prior nucleoside reverse transcriptase inhibitor (NRTI) exposure or who had HIV-1 isolates that contained multiple mutations conferring resistance to NRTIs had limited response to abacavir. The potential for cross-resistance between abacavir and other NRTIs should be considered when choosing new therapeutic regimens in therapy-experienced patients.
Related Products That Are Not Recommended
TRIZIVIR is a fixed-dose combination of 3 nucleoside analogue reverse transcriptase inhibitors (abacavir, lamivudine, and zidovudine). Concomitant administration of TRIZIVIR with other products containing abacavir, lamivudine, or zidovudine is not recommended. In addition, do not administer TRIZIVIR in combination with products containing emtricitabine.
Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Hypersensitivity Reaction
Inform patients
- that a Medication Guide and Warning Card summarizing the symptoms of the abacavir hypersensitivity reaction and other product information will be dispensed by the pharmacist with each new prescription and refill of TRIZIVIR, and instruct the patient to read the Medication Guide and Warning Card every time to obtain any new information that may be present about TRIZIVIR. The complete text of the Medication Guide is reprinted at the end of this document.
- to carry the Warning Card with them.
- how to identify a hypersensitivity reaction.
- that if they develop symptoms consistent with a hypersensitivity reaction they should call their healthcare provider right away to determine if they should stop taking TRIZIVIR.
- that a hypersensitivity reaction can worsen and lead to hospitalization or death if TRIZIVIR is not immediately discontinued.
- to not restart TRIZIVIR or any other abacavir-containing product following a hypersensitivity reaction because more severe symptoms can occur within hours and may include life-threatening hypotension and death.
- that a hypersensitivity reaction is usually reversible if it is detected promptly and TRIZIVIR is stopped right away.
- that if they have interrupted TRIZIVIR for reasons other than symptoms of hypersensitivity (for example, those who have an interruption in drug supply), a serious or fatal hypersensitivity reaction may occur with reintroduction of abacavir.
- to not restart TRIZIVIR or any other abacavir-containing product without medical consultation and only if medical care can be readily accessed by the patient or others.
Related Products that are Not Recommended
Inform patients that they should not take TRIZIVIR with ATRIPLA®, COMBIVIR, COMPLERA®, DUTREBIS™, EMTRIVA®, EPIVIR, EPIVIR-HBV®, EPZICOM® , RETROVIR, STRIBILD®, TRIUMEQ®, TRUVADA®, or ZIAGEN.
Neutropenia and Anemia
Inform patients that the important toxicities associated with zidovudine are neutropenia and/or anemia. Inform them of the extreme importance of having their blood counts followed closely while on therapy, especially for patients with advanced HIV-1 disease.
Myopathy
Inform patients that myopathy and myositis with pathological changes, similar to that produced by HIV-1 disease, have been associated with prolonged use of zidovudine.
Lactic Acidosis/Hepatomegaly
Inform patients that some HIV medicines, including TRIZIVIR, can cause a rare, but serious condition called lactic acidosis with liver enlargement (hepatomegaly).
Patients with Hepatitis B or C Co-infection
Advise patients co-infected with HIV-1 and HBV that worsening of liver disease has occurred in some cases when treatment with lamivudine was discontinued. Advise patients to discuss any changes in regimen with their physician.
Inform patients with HIV-1/HCV co-infection that hepatic decompensation (some fatal) has occurred in HIV-1/HCV co-infected patients receiving combination antiretroviral therapy for HIV-1 and interferon alfa with or without ribavirin.
Immune Reconstitution Syndrome
In some patients with advanced HIV infection, signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response, enabling the body to fight infections that may have been present with no obvious symptoms. Advise patients to inform their healthcare provider immediately of any symptoms of infection.
Redistribution/Accumulation of Body Fat
Inform patients that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time.
Information about HIV-1 Infection
TRIZIVIR is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients must remain on continuous HIV therapy to control HIV-1 infection and decrease HIV-related illness. Inform patients that sustained decreases in plasma HIV RNA have been associated with a reduced risk of progression to AIDS and death.
Advise patients to remain under the care of a physician when using TRIZIVIR. Advise patients to take all HIV medications exactly as prescribed. Advise patients to avoid doing things that can spread HIV-1 infection to others.
Advise patients not to re-use or share needles or other injection equipment.
Advise patients not to share personal items that can have blood or body fluids on them, like toothbrushes and razor blades.
Advise patients to always practice safer sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
Female patients should be advised not to breastfeed. Mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk.
Instruct patients that if they miss a dose, they should take it as soon as they remember. If they do not remember until it is time for the next dose, they should be instructed to skip the missed dose and go back to the regular schedule. Patients should not double their next dose or take more than the prescribed dose.
Instruct patients to read the Medication Guide before starting TRIZIVIR and to reread it each time the prescription is renewed. Instruct patients to inform their physician or pharmacist if they develop any unusual symptom, or if any known symptom persists or worsens.
Nonclinical Toxicology
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenicity
Abacavir: Abacavir was administered orally at 3 dosage levels to separate groups of mice and rats in 2-year carcinogenicity studies. Results showed an increase in the incidence of malignant and non-malignant tumors. Malignant tumors occurred in the preputial gland of males and the clitoral gland of females of both species, and in the liver of female rats. In addition, non-malignant tumors also occurred in the liver and thyroid gland of female rats. These observations were made at systemic exposures in the range of 6 to 32 times the human exposure at the recommended dose of 600 mg.
Lamivudine: Long-term carcinogenicity studies with lamivudine in mice and rats showed no evidence of carcinogenic potential at exposures up to 10 times (mice) and 58 times (rats) the human exposures at the recommended dose of 300 mg.
Zidovudine: Zidovudine was administered orally at 3 dosage levels to separate groups of mice and rats (60 females and 60 males in each group). Initial single daily doses were 30, 60, and 120 mg per kg per day in mice and 80, 220, and 600 mg per kg per day in rats. The doses in mice were reduced to 20, 30, and 40 mg per kg per day after day 90 because of treatment-related anemia, whereas in rats only the high dose was reduced to 450 mg per kg per day on day 91 and then to 300 mg per kg per day on day 279.
In mice, 7 late-appearing (after 19 months) vaginal neoplasms (5 nonmetastasizing squamous cell carcinomas, 1 squamous cell papilloma, and 1 squamous polyp) occurred in animals given the highest dose. One late-appearing squamous cell papilloma occurred in the vagina of a middle-dose animal. No vaginal tumors were found at the lowest dose.
In rats, 2 late-appearing (after 20 months), nonmetastasizing vaginal squamous cell carcinomas occurred in animals given the highest dose. No vaginal tumors occurred at the low or middle dose in rats. No other drug-related tumors were observed in either sex of either species.
At doses that produced tumors in mice and rats, the estimated drug exposure (as measured by AUC) was approximately 3 times (mouse) and 24 times (rat) the estimated human exposure at the recommended therapeutic dose of 100 mg every 4 hours.
It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
Two transplacental carcinogenicity studies were conducted in mice. One study administered zidovudine at doses of 20 mg per kg per day or 40 mg per kg per day from gestation day 10 through parturition and lactation with dosing continuing in offspring for 24 months postnatally. At these doses, exposures were approximately 3 times the estimated human exposure at the recommended doses. After 24 months at the 40-mg per kg per day dose, an increase in incidence of vaginal tumors was noted with no increase in tumors in the liver or lung or any other organ in either gender. These findings are consistent with results of the standard oral carcinogenicity study in mice, as described earlier. A second study administered zidovudine at maximum tolerated doses of 12.5 mg per day or 25 mg per day (approximately 1,000 mg per kg nonpregnant body weight or approximately 450 mg per kg of term body weight) to pregnant mice from days 12 through 18 of gestation. There was an increase in the number of tumors in the lung, liver, and female reproductive tracts in the offspring of mice receiving the higher dose level of zidovudine.
Mutagenicity
Abacavir: Abacavir induced chromosomal aberrations both in the presence and absence of metabolic activation in an in vitro cytogenetic study in human lymphocytes. Abacavir was mutagenic in the absence of metabolic activation, although it was not mutagenic in the presence of metabolic activation in an L5178Y mouse lymphoma assay. Abacavir was clastogenic in males and not clastogenic in females in an in vivo mouse bone marrow micronucleus assay. Abacavir was not mutagenic in bacterial mutagenicity assays in the presence and absence of metabolic activation.
Lamivudine: Lamivudine was mutagenic in an L5178Y mouse lymphoma assay and clastogenic in a cytogenetic assay using cultured human lymphocytes. Lamivudine was not mutagenic in a microbial mutagenicity assay, in an in vitro cell transformation assay, in a rat micronucleus test, in a rat bone marrow cytogenetic assay, and in an assay for unscheduled DNA synthesis in rat liver.
Zidovudine: Zidovudine was mutagenic in an L5178Y mouse lymphoma assay, positive in an in vitro cell transformation assay, clastogenic in a cytogenetic assay using cultured human lymphocytes, and positive in mouse and rat micronucleus tests after repeated doses. It was negative in a cytogenetic study in rats given a single dose.
Impairment of Fertility
Abacavir or Lamivudine: Abacavir or lamivudine did not affect male or female fertility in rats at a dose associated with exposures approximately 8 or 130 times, respectively, higher than the exposures in humans at the doses of 600 mg and 300 mg (respectively).
Zidovudine:
Use In Specific Populations
Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies of TRIZIVIR in pregnant women. Reproduction studies with abacavir, lamivudine, and zidovudine have been performed in animals (see Abacavir, Lamivudine, and Zidovudine sections below). TRIZIVIR should be used during pregnancy only if the potential benefits outweigh the risks.
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to TRIZIVIR during pregnancy. Physicians are encouraged to register patients by calling the Antiretroviral Pregnancy Registry at 1-800-258-4263.
Abacavir
Studies in pregnant rats showed that abacavir is transferred to the fetus through the placenta. Fetal malformations (increased incidences of fetal anasarca and skeletal malformations) and developmental toxicity (depressed fetal body weight and reduced crown-rump length) were observed in rats at a dose which produced 35 times the human exposure, based on AUC. Embryonic and fetal toxicities (increased resorptions, decreased fetal body weights) and toxicities to the offspring (increased incidence of stillbirth and lower body weights) occurred at half of the above-mentioned dose in separate fertility studies conducted in rats. In the rabbit, no developmental toxicity and no increases in fetal malformations occurred at doses that produced 8.5 times the human exposure at the recommended dose based on AUC.
Lamivudine
Studies in pregnant rats showed that lamivudine is transferred to the fetus through the placenta. Reproduction studies with orally administered lamivudine have been performed in rats and rabbits at doses producing plasma levels up to approximately 35 times that for the recommended adult HIV dose. No evidence of teratogenicity due to lamivudine was observed. Evidence of early embryolethality was seen in the rabbit at exposure levels similar to those observed in humans, but there was no indication of this effect in the rat at exposure levels up to 35 times those in humans.
Zidovudine
Reproduction studies with orally administered zidovudine in the rat and in the rabbit at doses up to 500 mg per kg per day revealed no evidence of teratogenicity with zidovudine. Zidovudine treatment resulted in embryo/fetal toxicity as evidenced by an increase in the incidence of fetal resorptions in rats given 150 or 450 mg per kg per day and rabbits given 500 mg per kg per day. The doses used in the teratology studies resulted in peak zidovudine plasma concentrations (after one-half of the daily dose) in rats 66 to 226 times, and in rabbits 12 to 87 times, mean steady-state peak human plasma concentrations (after one-sixth of the daily dose) achieved with the recommended daily dose (100 mg every 4 hours). In an additional teratology study in rats, a dose of 3,000 mg per kg per day (very near the oral median lethal dose in rats of approximately 3,700 mg per kg) caused marked maternal toxicity and an increase in the incidence of fetal malformations. This dose resulted in peak zidovudine plasma concentrations 350 times peak human plasma concentrations. No evidence of teratogenicity was seen in this experiment at doses of 600 mg per kg per day or less. Two rodent carcinogenicity studies were conducted.
Lactation
The Centers for Disease Control and Prevention recommend that HIV-1-infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection. Because of the potential for HIV-1 transmission mothers should be instructed not to breastfeed.
Pediatric Use
TRIZIVIR is not recommended in children who weigh less than 40 kg because it is a fixed-dose tablet that cannot be adjusted for these patient populations.
Therapy-Experienced Pediatric Trial
A randomized, double-blind trial, CNA3006, compared ZIAGEN plus lamivudine and zidovudine versus lamivudine and zidovudine in pediatric subjects, most of whom were extensively pretreated with nucleoside analogue antiretroviral agents. Subjects in this trial had a limited response to abacavir.
Geriatric Use
Clinical trials of abacavir, lamivudine, and zidovudine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, caution should be exercised in the administration of TRIZIVIR in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Patients With Impaired Renal Function
TRIZIVIR is not recommended for patients with creatinine clearance less than 50 mL per min because TRIZIVIR is a fixed-dose combination and the dosage of the individual components cannot be adjusted. If a dose reduction of the lamivudine or zidovudine components of TRIZIVIR is required for patients with renal impairment then the individual components should be used.
Patients With Impaired Hepatic Function
TRIZIVIR is a fixed-dose combination and the dosage of the individual components cannot be adjusted. If a dose reduction of abacavir, a component of TRIZIVIR, is required for patients with mild hepatic impairment (Child-Pugh Class A), then the individual components should be used.
The safety, efficacy, and pharmacokinetic properties of abacavir have not been established in patients with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment; therefore, TRIZIVIR is contraindicated in these patients.
Zidovudine is primarily eliminated by hepatic metabolism and zidovudine concentrations are increased in patients with impaired hepatic function, which may increase the risk of hematologic toxicity. Frequent monitoring of hematologic toxicities is advised.
記載されている薬物相互作用試験は、 単一のエンティティとしてのアバカビル、ラミブジンまたはジドブジン;薬物相互作用はありません。 TRIZIVIRを使用して試験が行われました。臨床的に重要な薬物はありません。 アバカビル、ラミブジン、ジドブジンの間で相互作用が予想されます。.
チトクロームP450酵素。: アバカビル、ラミブジン、そして。 ジドブジンはチトクロームP450酵素によって有意に代謝されません。 したがって、臨床的に重要な薬物相互作用が起こる可能性は低いです。 これらの経路を通じて代謝される薬物で発生します。.
グルクロニルトランスフェラーゼ。: 共通のため。 グルクロニルトランスフェラーゼを介したアバカビルとジドブジンの代謝経路、15。 HIV-1に感染した被験者は、シングルを評価するクロスオーバー試験に登録されました。 アバカビル(600 mg)、ラミブジン(150 mg)、およびジドブジン(300 mg)のみの用量。 または組み合わせて。. 分析では、臨床的に関連する変化は見られませんでした。 ラミブジンまたはジドブジンの添加によるアバカビルの薬物動態または。 ラミブジンとジドブジンの組み合わせ。. ラミブジン暴露(AUC。 15%減少)およびジドブジン曝露(AUC増加10%)は示さなかった。 同時アバカビルによる臨床的に関連する変化。.
その他の相互作用。
エタノール。: アバカビルはに影響を与えません。 エタノールの薬物動態特性。. エタノールはの排除を減らします。 アバカビルは全体的な曝露の増加を引き起こします。.
インターフェロンアルファ。: 重要はありませんでした。 試験におけるラミブジンとインターフェロンアルファ間の薬物動態学的相互作用。 19人の健康な男性被験者の。.
メタドン。: 11人のHIV-1感染者の試験で。 メタドン維持療法を受けている被験者(毎日40 mgおよび90 mg)。 600 mgのアバカビル1日2回(現在推奨されている用量の2倍)、経口。 メタドンクリアランスは22%増加しました(90%CI:6%から42%)。. メタドンの添加は、臨床的に有意な影響を与えません。 アバカビルの薬物動態特性。.
リバビリン。: In vitro。 データはリバビリンを示しています。 ラミブジン、スタブジン、ジドブジンのリン酸化を減らします。. ただし、違います。 薬物動態(例:.、血漿濃度または細胞内三リン酸。 活性代謝物濃度)または薬力学的(例:.、HIV-1 / HCVの喪失。 ウイルス学的抑制)相互作用は、リバビリンとラミブジンのときに観察されました。 (n = 18)、スタブジン(n = 10)、またはジドブジン(n = 6)を一部として同時投与しました。 HIV-1 / HCV同時感染被験者への多剤レジメンの。.
アバカビルに対する他の同時投与薬物の影響。 ラミブジン、またはジドブジンを表4に示します。.
表4:アバカビルに対する同時投与薬物の影響。
ラミブジン、およびジドブジンAUC。a
| 薬物と用量の同時投与。 | 薬物と用量。 | n | アバカビル、ラミブジン、またはジドブジンの濃度。 | 併用薬の濃度。 | |
| AUC。 | 変動。 | ||||
| エタノール0.7 g / kg。 | アバカビルシングル600 mg。 | 24 | ⁇ 41%。 | 90%CI:35%から48%。 | ↔b |
| ネルフィナビル750 mg 8時間x 7〜10日ごと。 | ラミブジンシングル150 mg。 | 11 | ⁇ 10%。 | 95%CI:1%から20%。 | ↔ |
| トリメトプリム160 mg /スルファメトキサゾール800 mg 1日x 5日。 | ラミブジンシングル300 mg。 | 14 | ⁇ 43%。 | 90%CI:32%から55%。 | ↔ |
| アトバコン750 mgを12時間ごとに食物と一緒に。 | ジドブジン200 mg 8時間ごと。 | 14 | ⁇ 31%。 | 範囲:23%から78%。c | ↔ |
| クラリスロマイシン500 mgを1日2回。 | ジドブジン100 mg 4時間x 7日ごと。 | 4 | ⁇ 12%。 | 範囲: ⁇ 34%から ⁇ 14%。 | 報告されていません。 |
| フルコナゾール400 mg毎日。 | ジドブジン200 mg 8時間ごと。 | 12 | ⁇ 74%。 | 95%CI:54%から98%。 | 報告されていません。 |
| メタドン30〜90 mg毎日。 | ジドブジン200 mg 4時間ごと。 | 9 | ⁇ 43%。 | 範囲:16%から64%c。 | ↔ |
| ネルフィナビル750 mg 8時間x 7〜10日ごと。 | ジドブジンシングル200 mg。 | 11 | ⁇ 35%。 | 範囲:28%から41%。 | ↔ |
| プロベネシド500 mg 6時間x 2日ごと。 | ジドブジン2 mg / kg 8時間x 3日ごと。 | 3 | ⁇ 106%。 | 範囲:100%から170%。c | 評価されていません。 |
| リファンピン600 mg毎日x 14日。 | ジドブジン200 mg 8時間x 14日ごと。 | 8 | ⁇ 47%。 | 90%CI:41%から53%。 | 評価されていません。 |
| リトナビル300 mg 6時間x 4日ごと。 | ジドブジン200 mg 8時間x 4日ごと。 | 9 | ⁇ 25%。 | 95%CI:15%から34%。 | ↔ |
| バルプロ酸250 mgまたは500 mg 8時間x 4日ごと。 | ジドブジン100 mg 8時間x 4日ごと。 | 6 | ⁇ 80%。 | 範囲:64%から130%。c | 評価されていません。 |
| ⁇ =増加; ⁇ =減少; ⁇ =いいえ。
大幅な変更。 AUC =濃度対時間曲線の下の領域。 CI =。
信頼区間。. a見る。 薬物相互作用。 薬物に関する追加情報。 相互作用。. b薬物間相互作用は男性でのみ評価されました。. cパーセント差の推定範囲。. |
妊娠カテゴリーC
適切で適切に管理された研究はありません。 妊娠中の女性のトリジビル。. アバカビル、ラミブジン、および ジドブジンは動物で実施されています(参照)。 アバカビル、ラミブジン、そして。 以下のジドブジンセクション。)。. TRIZIVIRは妊娠中のみ使用してください。 潜在的な利益がリスクを上回る場合。.
妊娠暴露登録。
監視する妊娠暴露登録があります。 妊娠中のTRIZIVIRに曝露した女性の妊娠転帰。. 医師。 抗レトロウイルス妊娠を呼び出して患者を登録することをお勧めします。 1-800-258-4263のレジストリ。.
アバカビル。
妊娠中のラットを用いた研究では、アバカビルが 胎盤を介して胎児に移されました。. 胎児奇形(増加。 胎児の ⁇ 門と骨格奇形の発生)および発達。 毒性(胎児の体重の減少とクラウンランプの長さの減少)があった。 に基づいて、ヒトへの暴露の35倍を生成する用量でラットで観察された。 AUC。胚毒性と胎児毒性(吸収の増加、胎児の減少)。 体重)と子孫への毒性(発生率の増加)。 死産と体重減少)は、上記の用量の半分で発生しました。 ラットで行われた別の受胎能研究で。. ウサギでは、いいえ。 発生毒性および胎児奇形の増加はありませんでした。 推奨用量ベースでヒトへの暴露の8.5倍をもたらした用量。 AUCで.
ラミブジン。
妊娠中のラットを用いた研究では、ラミブジンがそうであることが示されました。 胎盤を介して胎児に移されました。. 経口での生殖研究。 ラミブジンの投与は、ラットとウサギの用量で行われた。 推奨のプラズマレベルの約35倍までの血漿レベルを生成します。 成人のHIV投与量。. ラミブジンによる催奇形性の証拠は観察されなかった。. 初期の胚発生の証拠は、曝露レベルでウサギに見られました。 ヒトで観察されたものと同様ですが、この影響の兆候はありませんでした。 ヒトの35倍までの暴露レベルでラットに。.
ジドブジン。
経口投与されたジドブジンを用いた生殖試験。 ラットおよびウサギでは、1日あたり1 kgあたり500 mgまでの用量で、明らかにされなかった。 ジドブジンによる催奇形性の証拠。. ジドブジン治療がもたらされました。 胎児の発生率の増加によって証明される胚/胎児毒性。 1日あたり1 kgあたり150または450 mgを投与されたラットと500 mgを投与されたウサギの吸収。 1日あたりkgあたり。. 奇形学研究で使用された用量はピークをもたらしました。 ラットにおけるジドブジンの血漿濃度(1日量の半分後)66。 226回まで、ウサギでは12〜87回、定常状態のピーク人間を意味します。 血漿濃度(1日量の6分の1後)は、 推奨される1日量(4時間ごとに100 mg)。. 追加の奇形学研究で。 ラットでは、1日あたり1 kgあたり3,000 mgの用量(経口中央値致死量に非常に近い)。 ラットの用量は約3,700 mg / kg)で、顕著な母体毒性を引き起こした。 胎児奇形の発生率の増加。. この線量は結果となった。 ピークジドブジン血漿濃度ピークヒト血漿の350倍。 濃度。. この実験では催奇形性の証拠は見られなかった。 1日あたりkgあたり600mg以下の用量。. 2つのげっ歯類発がん性試験があった。 実施。.
以下の副作用については、他にも説明します。 ラベルのセクション:。
- 重 ⁇ で、時には致命的な過敏反応。.
- 好中球減少症や貧血を含む血液毒性。.
- 症候性ミオパシー。.
- 乳酸アシドーシスと脂肪症を伴う重度の肝腫大。.
- B型肝炎の悪化
- HIV-1に同時感染した患者の肝代償不全。 C型肝炎
- HIV-1 / HCV同時感染患者における貧血の悪化。 リバビリンとジドブジンの投与。.
- 免疫再構成症候群。.
- 脂肪の再分配。.
- 心筋 ⁇ 塞。.
臨床試験の経験。
臨床試験は広く行われているためです。 さまざまな条件、aの臨床試験で観察された副作用率。 薬物は、別の臨床試験の率と直接比較することはできません。 薬物であり、臨床診療で観察された率を反映していない可能性があります。.
重 ⁇ で致命的なアバカビル関連過敏症。 反応。
臨床試験では、深刻で、時には致命的です。 過敏反応は、トリジビルの成分であるアバカビルで発生しました。 これらの反応は2つ以上特徴付けられています。.以下の兆候または症状の:(1)発熱; (2)発疹; (3)消化器。 症状(吐き気、 ⁇ 吐、下 ⁇ 、腹痛など)。 (4)。 体質症状(全身 ⁇ 怠感、疲労感、または痛みを含む);。 (5)呼吸器症状(呼吸困難、咳、 ⁇ 頭炎を含む)。. ほとんどすべて。 アバカビル過敏反応には、発熱や発疹などがあります。 症候群。.
その他の兆候や症状には、 ⁇ 眠、頭痛などがあります。 筋肉痛、浮腫、関節痛、感覚異常。. アナフィラキシー、肝不全、腎臓。 失敗、低血圧、成人呼吸 ⁇ 迫症候群、呼吸不全、 筋融解症、および死はこれらの過敏症に関連して発生しました。 反応。. 身体的所見には、リンパ節腫 ⁇ 、粘膜が含まれています。 病変(結膜炎および口内 ⁇ 瘍)、黄斑丘疹またはじんま疹。 発疹(一部の患者は他の種類の発疹があり、他の患者は発疹がありませんでした。 発疹)。. 多形紅斑の報告がありました。. 実験室の異常。 肝化学の上昇、クレアチンホスホキナーゼの上昇、上昇。 クレアチニン、リンパ球減少症、異常な胸部X線所見(主に ローカライズされた浸透)。.
TRIZIVIRを使用した追加の副作用。
治療に伴う臨床的副作用(評価者。 中等度または重度としての研究者)、頻度以上。 アバカビル300 mgを1日2回、ラミブジン150 mgを2回併用して、治療中に5%まで。 1日2回ジドブジン300 mgをインディナビル800 mgと比較して3回。 毎日、ラミブジン150 mgを1日2回、ジドブジン300 mgを1日2回から。 CNA3005を表1に示します。.
表1:治療-緊急(すべての因果関係)有害。
少なくとも中程度の強度での反応(グレード2〜4、それ以上または同等。
5%の頻度)治療歴のある成人(CNA3005)で48週間の治療。
| 副作用。 | ZIAGENとラミブジン/ジドブジン。 (n = 262)。 |
インディナビルとラミブジン/ジドブジン。 (n = 264)。 |
| 吐き気。 | 19%。 | 17%。 |
| 頭痛。 | 13%。 | 9% |
| ⁇ 怠感と疲労。 | 12%。 | 12%。 |
| 吐き気と ⁇ 吐。 | 10%。 | 10%。 |
| 過敏反応。 | 8% | 2% |
| 下 ⁇ 。 | 7% | 5% |
| 発熱および/または悪寒。 | 6% | 3% |
| うつ病性障害。 | 6% | 4% |
| 筋骨格痛。 | 5% | 7% |
| 皮膚の発疹。 | 5% | 4% |
| 耳/鼻/喉の感染症。 | 5% | 4% |
| ウイルス性呼吸器感染症。 | 5% | 5% |
| 不安。 | 5% | 3% |
| 腎徴候/症状。 | <1%。 | 5% |
| 痛み(非サイト固有)。 | <1%。 | 5% |
5人の被験者が受けています。 CNA3005のアバカビルは、既存のうつ病と比較して悪化を経験しました。 インディナビル群の誰にも。. 既存のうつ病のバックグラウンド率。 2つの治療群で類似していた。.
実験室の異常。
実験室異常。 CNA3005を表2に示します。.
表2:治療緊急。
CNA3005の検査異常(グレード3/4)。
| 実験パラメータ。 | ZIAGENとラミブジン/ジドブジン。 (n = 262)。 |
インディナビルとラミブジン/ジドブジン。 (n = 264)。 |
| CPKの上昇(> 4 x ULN)。 | 18(7%)。 | 18(7%)。 |
| ALT(> 5.0 x ULN)。 | 16(6%)。 | 16(6%)。 |
| 好中球減少症(<750 /mm³)。 | 13(5%)。 | 13(5%)。 |
| 高トリグリセリド血症(> 750 mg / dL)。 | 5(2%)。 | 3(1%)。 |
| 高アミラ血症(> 2.0 x ULN)。 | 5(2%)。 | 1(<1%)。 |
| 高血糖(> 13.9 mmol / L)。 | 2(<1%)。 | 2(<1%)。 |
| 貧血(Hgb≤6.9 g / dL)。 | 0(0%)。 | 3(1%)。 |
| ULN =通常の上限。. n =評価された被験者の数。. |
その他の有害事象。
逆行に加えて。 表1および2の反応、拡大で観察された他の有害事象。 アバカビルのアクセスプログラムは ⁇ 炎とGGTの増加でした。
市販後の経験。
以下の副作用。 市販後の使用中に確認されています。. これらの反応があるからです。 未知のサイズの人口から自発的に報告され、常にではありません。 それらの頻度を確実に推定するか、因果関係を確立することが可能です。 薬物暴露に。.
アバカビル。
心血管:。 心筋 ⁇ 塞。.
皮膚:。 スティーブンス・ジョンソン症候群(SJS)の疑い。 毒性の表皮壊死症(TEN)は、投与されている患者で報告されています。 アバカビルは、主に関連することが知られている薬と組み合わせて。 SJSとTEN、それぞれ。. 臨床徴候の重複のため。 アバカビルとSJSとTENに対する過敏症の間の症状、および 一部の患者では複数の薬物感受性の可能性、アバカビルはそうあるべきです。 そのような場合は中止され、再開されません。. 報告もありました。 アバカビルを使用した多形紅斑。.
アバカビル、ラミブジン、および/またはジドブジン。
全体としての体:。 再配布/蓄積。 体脂肪。.
心血管:。 心筋症。.
消化器:。 口内炎。.
内分 ⁇ と代謝:。 女性化乳房。.
消化管:。 拒食症および/または減少。 食欲、腹痛、消化不良、口腔粘膜色素沈着。.
一般:。 血管炎、脱力感。.
貧血とリンパ:。 再生不良性貧血、貧血。 (純粋な赤血球無形成症および治療中に進行する重度の貧血を含む)、。 リンパ節腫 ⁇ 、 ⁇ 腫、血小板減少症。.
肝臓:。 乳酸アシドーシスと肝脂肪症、上昇。 ビリルビン、トランスアミナーゼの上昇、B型肝炎の治療後の悪化
過敏症:。 感作反応。 (アナフィラキシーを含む)、じんま疹。.
筋骨格:。 関節痛、筋肉痛、筋肉。 脱力感、横紋筋融解症。. 神経質:めまい、感覚異常、末 ⁇ 。 神経障害、発作。.
精神医学:。 不眠症およびその他の睡眠障害。. 呼吸器:。 異常な呼吸音/ ⁇ 鳴。. 皮膚:脱毛症、多形紅斑、 スティーブンス・ジョンソン症候群。.
過剰摂取に対する既知の特定の治療法はありません。 トリジビル。過剰摂取が発生した場合、患者を監視し、標準にする必要があります。 必要に応じてサポート処理を適用します。.
アバカビル。
アバカビルを取り除くことができるかどうかは不明です。 腹膜透析または血液透析。.
ラミブジン。
無視できる量のラミブジンが除去されたためです。 (4時間)血液透析、継続的な外来腹膜透析、および自動化。 腹膜透析、継続的な血液透析が提供されるかどうかは不明です。 ラミブジンの過剰摂取イベントにおける臨床的利益。.
ジドブジン。
ジドブジンの急性過剰摂取が報告されています。 小児患者と成人。. これらには、最大50グラムの曝露が含まれていました。. 番号。 特定の症状または兆候は、急性過剰摂取後に確認されています。 疲労、頭痛などの有害事象としてリストされているもの以外のジドブジン。 ⁇ 吐、および血液学的障害の不定期の報告。. 患者。 永久的な後遺症なしで回復した。. 血液透析と腹膜透析。 ジドブジンの除去に無視できるほどの影響があるようです。 その主要代謝物である3'azido-3'-deoxy-5'-O-β-D-glucopyranuronosylthymidineの排除。 (GZDV)、拡張されています。.
成人の薬物動態。
単回投与の3ウェイクロスオーバーバイオアベイラビリティ試験。 1つのTRIZIVIRタブレットと1つのZIAGENタブレット(300 mg)、1つのEPIVIRタブレット(150 mg)の 加えて、健康な被験者に同時に投与される1つのRETROVIRタブレット(300 mg)。 (n = 24)、測定した吸収の程度に違いはありませんでした。 血漿濃度-時間曲線(AUC)の下の領域と最大ピーク。 3つの成分すべての濃度(Cmax)。. 1つのTRIZIVIRタブレットでした。 1つのZIAGENタブレット(300 mg)、1つのEPIVIRタブレット(150 mg)、および1。 空腹時への単回投与後のレトロビル錠(300 mg)。 健康な被験者(n = 24)。.
アバカビル。: 経口投与後、アバカビル。 急速に吸収され、広範囲に分布しています。. の経口投与後。 20人の被験者で1日2回300 mgのアバカビル、Cmaxは1 mLあたり3.0±0.89 mcgでした。 (平均±SD)およびAUC(0-12時間)は、6.02±1.73 mcg•mLあたりの時間でした。の結合。 ヒト血漿タンパク質へのアバカビルは約50%であり、独立していた。 濃度。. 総血中および血漿中薬物関連放射能濃度。 同一であり、アバカビルが容易に分布することを示しています。 赤血球。. アバカビルの主要な排 ⁇ 経路は代謝です。 アルコールデヒドロゲナーゼは5'-カルボン酸とグルクロニルを形成します。 5'-グルクロニドを形成するトランスフェラーゼ。.
ラミブジン。: 経口投与後、 ラミブジンは急速に吸収され、広範囲に分布します。. プラズマに結合します。 タンパク質は低いです。. ラミブジンの静脈内投与の約70%です。 尿中の未変化の薬物として回復した。. ラミブジンの代謝はマイナーです。 排除のルート。. 人間では、唯一知られている代謝物はです。 トランススルホキシド代謝物(12時間後の経口投与量の約5%)。.
ジドブジン。: 経口投与後、 ジドブジンは急速に吸収され、広範囲に分布しています。. プラズマに結合します。 タンパク質は低いです。. ジドブジンは主に肝代謝によって排除されます。. 。 ジドブジンの主要代謝産物はGZDVです。 GZDV AUCは約3倍です。 ジドブジンAUC。ジドブジンとGZDVの尿中回復は14%を占めます。 経口投与後の用量のそれぞれ74%。. 一瞬。 代謝物、3'-アミノ-3'デオキシチミジン(AMT)が確認されています。 プラズマ。. AMT AUCはジドブジンAUCの5分の1でした。
ヒトでは、アバカビル、ラミブジン、ジドブジンはそうではありません。 シトクロムP450酵素によって著しく代謝されます。.
アバカビル、ラミブジンの薬物動態特性。 空腹時の被験者のジドブジンを表3にまとめます。.
表3:薬物動態パラメータ。a ために。
成人のアバカビル、ラミブジン、ジドブジン。
| パラメータ。 | アバカビル。 | ラミブジン。 | ジドブジン。 | |||
| 経口バイオアベイラビリティ(%)。 | 86±25。 | n = 6。 | 86±16。 | n = 12。 | 64±10。 | n = 5。 |
| 見かけの分布量(L / kg)。 | 0.86±0.15。 | n = 6。 | 1.3±0.4。 | n = 20。 | 1.6±0.6。 | n = 8。 |
| 全身クリアランス(L / h / kg)。 | 0.80±0.24。 | n = 6。 | 0.33±0.06。 | n = 20。 | 1.6±0.6。 | n = 6。 |
| 腎クリアランス(L / h / kg)。 | 0.007±0.008。 | n = 6。 | 0.22±0.06。 | n = 20。 | 0.34±0.05。 | n = 9。 |
| 消失半減期(h)。 | 1.45±0.32。 | n = 20。 | 5から7。b | 0.5から3。b | ||
| aデータは平均±標準偏差として表示されます。
記載されている場合を除きます。. bおおよその範囲。. |
TRIZIVIRの吸収に対する食品の影響。
単回投与のバイオアベイラビリティにおける食物による投与。 試験の結果、以前に観察された結果と同様に、Cmaxが低下しました。 参照定式化。. アバカビル、ラミブジンの平均[90%CI]減少。 ジドブジンCmaxは32%[24%〜38%]、18%[10%〜25%]、28%[13%〜40%]でした。 と比較して、高脂肪食と一緒に投与された場合。 空腹時の投与。. 食物によるトリジビルの投与。 アバカビル、ラミブジン、ジドブジンの吸収の程度は変わりませんでした。 (AUC)、空腹時の投与と比較(n = 24)。.
参照:
国で利用可能
 Australia
Australia Austria
Austria Belgium
Belgium Bulgaria
Bulgaria Canada
Canada China
China Croatia (Hrvatska)
Croatia (Hrvatska) Cyprus
Cyprus Czech Republic
Czech Republic Denmark
Denmark Ecuador
Ecuador Estonia
Estonia Finland
Finland France
France Georgia
Georgia Germany
Germany Greece
Greece Hong Kong
Hong Kong Hungary
Hungary Iceland
Iceland Ireland
Ireland Israel
Israel Italy
Italy Latvia
Latvia Liechtenstein
Liechtenstein Lithuania
Lithuania Luxembourg
Luxembourg Malta
Malta Netherlands
Netherlands Norway
Norway Oman
Oman Peru
Peru Poland
Poland Portugal
Portugal Romania
Romania Russia
Russia Serbia
Serbia Slovakia
Slovakia Slovenia
Slovenia Spain
Spain Sweden
Sweden Switzerland
Switzerland Taiwan
Taiwan United Kingdom
United Kingdom USA
USA