


Composición:
Solicitud:
Usado en tratamiento:
Revisión médica por Oliinyk Elizabeth Ivanovna Última actualización de farmacia el 10.04.2022

¡Atención! ¡La información en la página es solo para profesionales médicos! ¡La información se recopila en Fuentes abiertas y puede contener errores significativos! ¡Tenga cuidado y vuelva a verificar toda la información de esta página!
Los 20 mejores medicamentos con los mismos ingredientes:
Formas de dosificación y fortalezas
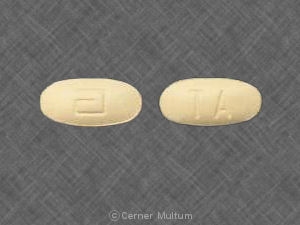
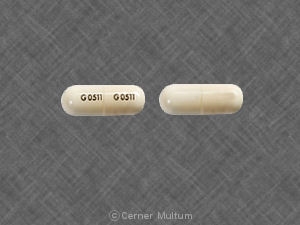
- 54 mg comprimidos amarillos, impresos con el logotipo "a" y las letras de identificación del código "TA".
- Comprimidos blancos de 160 mg, impresos con el logotipo "a" y las letras de identificación del código "TC".
Almacenamiento y manejo
TRICOR® (tabletas de fenofibrato) está disponible en dos puntos fuertes:
54 mg tabletas amarillas, impresas con el logotipo "a" y las letras de identificación de código "TA", disponibles en botellas de 90 (NDC 0074-4009-90).
160 mg tabletas blancas, impresas con el logotipo "a" y las letras de identificación de código "TC", disponibles en botellas de 90 (NDC 0074-4013-90).
Almacenamiento
Almacenar a 25 ° C (77 ° F); excursiones permitidas a 15-30 ° C (59-86 ° F).
Mantener fuera del alcance de los niños. Proteger de la humedad.
Fabricado por: Laboratoires Fournier, S.A., 21300 Chen´ve, Francia. Revisado: enero de 2017.
Hipercolesterolemia primaria o dislipidemia mixta
TRICOR está indicado como terapia complementaria a la dieta para reducir el colesterol elevado de lipoproteínas de baja densidad (LDL-C), el colesterol total (Total-C), los triglicéridos y la apolipoproteína B (Apo B), y para aumentar el colesterol de lipoproteínas de alta densidad (HDL-C) en pacientes adultos con hipercolesterolemia primaria o dislipidemia mixta.
Hipertrigliceridemia severa
TRICOR también está indicado como terapia complementaria a la dieta para el tratamiento de pacientes adultos con hipertrigliceridemia severa. Mejorar el control glucémico en pacientes diabéticos que muestran quilomicronemia en ayunas generalmente obviará la necesidad de intervención farmacológica.
Niveles marcadamente elevados de triglicéridos séricos (p. Ej. > 2,000 mg / dL) puede aumentar el riesgo de desarrollar pancreatitis. El efecto de la terapia con fenofibrato en la reducción de este riesgo no se ha estudiado adecuadamente.
Importantes limitaciones de uso
No se demostró que el fenofibrato a una dosis equivalente a 160 mg de TRICOR reduzca la morbilidad y mortalidad por enfermedad coronaria en un ensayo controlado aleatorio grande de pacientes con diabetes mellitus tipo 2.
Consideraciones generales
Los pacientes deben seguir una dieta adecuada para reducir los lípidos antes de recibir TRICOR, y deben continuar esta dieta durante el tratamiento con TRICOR. Las tabletas TRICOR deben administrarse con las comidas, optimizando así la biodisponibilidad del medicamento.
El tratamiento inicial para la dislipidemia es una terapia dietética específica para el tipo de anomalía de la lipoproteína. El exceso de peso corporal y el exceso de ingesta alcohólica pueden ser factores importantes en la hipertrigliceridemia y deben abordarse antes de cualquier terapia farmacológica. El ejercicio físico puede ser una medida auxiliar importante. Las enfermedades que contribuyen a la hiperlipidemia, como el hipotiroidismo o la diabetes mellitus, deben buscarse y tratarse adecuadamente. La terapia con estrógenos, los diuréticos tiazídicos y los betabloqueantes, a veces se asocian con aumentos masivos en los triglicéridos plasmáticos, especialmente en sujetos con hipertrigliceridemia familiar. En tales casos, la interrupción del agente etiológico específico puede obviar la necesidad de una terapia farmacológica específica de hipertrigliceridemia.
Los niveles de lípidos deben controlarse periódicamente y se debe considerar reducir la dosis de TRICOR si los niveles de lípidos caen significativamente por debajo del rango objetivo.
La terapia debe retirarse en pacientes que no tienen una respuesta adecuada después de dos meses de tratamiento con la dosis máxima recomendada de 160 mg una vez al día.
Hipercolesterolemia primaria o dislipidemia mixta
La dosis inicial de TRICOR es de 160 mg una vez al día.
Hipertrigliceridemia severa
La dosis inicial es de 54 a 160 mg por día. La dosis debe individualizarse de acuerdo con la respuesta del paciente y debe ajustarse si es necesario después de las determinaciones repetidas de lípidos a intervalos de 4 a 8 semanas. La dosis máxima es de 160 mg una vez al día.
Función renal deteriorada
El tratamiento con TRICOR debe iniciarse con una dosis de 54 mg por día en pacientes con insuficiencia renal leve a moderada, y aumentar solo después de la evaluación de los efectos sobre la función renal y los niveles de lípidos a esta dosis. Se debe evitar el uso de TRICOR en pacientes con insuficiencia renal grave.
Pacientes geriátricos
La selección de dosis para los ancianos debe realizarse en función de la función renal.
TRICOR está contraindicado en :
- pacientes con insuficiencia renal grave, incluidos los que reciben diálisis.
- pacientes con enfermedad hepática activa, incluidos aquellos con cirrosis biliar primaria y anormalidades persistentes inexplicables de la función hepática.
- pacientes con enfermedad preexistente de la vesícula biliar.
- madres lactantes
- pacientes con hipersensibilidad conocida al fenofibrato o al ácido fenofibrico.
ADVERTENCIAS
Incluido como parte de la "PRECAUCIONES" Sección
PRECAUCIONES
Mortalidad y morbilidad coronaria de la enfermedad cardíaca
No se ha establecido el efecto de TRICOR sobre la morbilidad y mortalidad por enfermedad coronaria y la mortalidad no cardiovascular.
El ensayo Acción para controlar el riesgo cardiovascular en la diabetes lipídica (ACCORD Lipid) fue un estudio aleatorizado controlado con placebo de 5518 pacientes con diabetes mellitus tipo 2 en terapia con estatina de fondo tratada con fenofibrato. La duración media del seguimiento fue de 4,7 años. La terapia combinada de fenofibrato más estatina mostró una reducción no significativa del riesgo relativo del 8% en el resultado primario de eventos cardiovasculares adversos importantes (MACE), un compuesto de infarto de miocardio no fatal, accidente cerebrovascular no mortal y muerte por enfermedad cardiovascular (razón de riesgo [HR ] 0.92, IC del 95% 0.79-1.08) (p =. En un análisis de subgrupos de género, la razón de riesgo para MACE en hombres que reciben terapia combinada versus monoterapia con estatina fue de 0.82 (IC 95% 0.69-0.99), y la razón de riesgo para MACE en mujeres que reciben terapia combinada versus monoterapia con estatina fue de 1.38 (IC 95% 0.98-1.94) (interacción p = 01). La importancia clínica de este hallazgo de subgrupo no está clara.
El estudio de intervención de fenofibrato y disminución de eventos en la diabetes (FIELD) fue un estudio aleatorizado de 5 años, controlado con placebo, de 9795 pacientes con diabetes mellitus tipo 2 tratados con fenofibrato. El fenofibrato demostró una reducción relativa no significativa del 11% en el resultado primario de eventos de enfermedad coronaria (razón de riesgo [HR] 0.89, IC del 95% 0.75-1.05, p = 0.16) y una reducción significativa del 11% en el resultado secundario de la enfermedad cardiovascular total eventos (HR 0.89 [0.80. Hubo un aumento no significativo del 11% (HR 1.11 [0.95, 1.29], p = 0.18) y del 19% (HR 1.19 [0.90, 1.57], p = 0.22) en la mortalidad total y coronaria de la enfermedad cardíaca, respectivamente, con fenofibrato en comparación con placebo.
Debido a las similitudes químicas, farmacológicas y clínicas entre TRICOR (tabletas de fenofibrato), clofibrato y gemfibrozilo, los hallazgos adversos en 4 grandes estudios clínicos aleatorizados controlados con placebo con estos otros fármacos de fibrato también pueden aplicarse a TRICOR
En el Proyecto de Medicamentos Coronarios, un gran estudio del infarto de miocardio posterior de pacientes tratados durante 5 años con clofibrato, no hubo diferencias en la mortalidad observada entre el grupo de clofibrato y el grupo de placebo. Sin embargo, hubo una diferencia en la tasa de colelitiasis y colecistitis que requieren cirugía entre los dos grupos (3.0% vs. 1.8%).
En un estudio realizado por la Organización Mundial de la Salud (OMS), 5000 sujetos sin enfermedad coronaria conocida fueron tratados con placebo o clofibrato durante 5 años y seguidos durante un año adicional. Hubo una mortalidad por todas las causas ajustada estadísticamente significativa y de mayor edad en el grupo de clofibrato en comparación con el grupo placebo (5.70% vs. 3.96%, p = <0.01). El exceso de mortalidad se debió a un aumento del 33% en las causas no cardiovasculares, incluida la malignidad, las complicaciones posteriores a la colistectomía y la pancreatitis. Esto pareció confirmar el mayor riesgo de enfermedad de la vesícula biliar observado en pacientes tratados con clofibrato estudiados en el Proyecto de Medicamentos Coronarios.
El Helsinki Heart Study fue un gran estudio (n = 4081) de hombres de mediana edad sin antecedentes de enfermedad de las arterias coronarias. Los sujetos recibieron placebo o gemfibrozilo durante 5 años, con una extensión abierta de 3.5 años después. La mortalidad total fue numéricamente mayor en el grupo de aleatorización de gemfibrozilo, pero no alcanzó significación estadística (p = 0.19, intervalo de confianza del 95% para el riesgo relativo G: P = .91-1.64). Aunque las muertes por cáncer tuvieron una tendencia más alta en el grupo de gemfibrozilo (p = 0.11), los cánceres (excluyendo el carcinoma de células basales) se diagnosticaron con la misma frecuencia en ambos grupos de estudio. Debido al tamaño limitado del estudio, no se demostró que el riesgo relativo de muerte por cualquier causa sea diferente al observado en los datos de seguimiento de 9 años del estudio de la Organización Mundial de la Salud (RR = 1.29).
Un componente de prevención secundaria del Helsinki Heart Study inscribió a hombres de mediana edad excluidos del estudio de prevención primaria debido a una enfermedad coronaria conocida o sospechada. Los sujetos recibieron gemfibrozilo o placebo durante 5 años. Aunque las muertes cardíacas tuvieron una tendencia más alta en el grupo gemfibrozilo, esto no fue estadísticamente significativo (razón de riesgo 2.2, intervalo de confianza del 95%: 0.94-5.05). La tasa de cirugía de vesícula biliar no fue estadísticamente significativa entre los grupos de estudio, pero sí tuvo una tendencia más alta en el grupo de gemfibrozilo (1.9% vs. 0.3%, p = 0.07).
Músculo esquelético
Las fibras aumentan el riesgo de miopatía y se han asociado con rabdomiólisis. El riesgo de toxicidad muscular grave parece aumentar en pacientes de edad avanzada y en pacientes con diabetes, insuficiencia renal o hipotiroidismo.
La miopatía debe considerarse en cualquier paciente con mialgias difusas, sensibilidad o debilidad muscular y / o elevaciones marcadas de los niveles de creatina fosfoquinasa (CPK).
Se debe aconsejar a los pacientes que informen de inmediato el dolor muscular inexplicable, la sensibilidad o la debilidad, particularmente si van acompañados de malestar o fiebre. Los niveles de CPK deben evaluarse en pacientes que informan estos síntomas, y la terapia TRICOR debe suspenderse si se producen niveles de CPK marcadamente elevados o se sospecha o diagnostica miopatía / miositis.
Los datos de estudios observacionales indican que el riesgo de rabdomiólisis aumenta cuando los fibratos, en particular el gemfibrozilo, se administran conjuntamente con un inhibidor de la HMG-CoA reductasa (estatina). La combinación debe evitarse a menos que el beneficio de nuevas alteraciones en los niveles de lípidos supere el mayor riesgo de esta combinación de medicamentos.
Se han notificado casos de miopatía, incluida la rabdomiólisis, con fenofibratos administrados conjuntamente con colchicina, y se debe tener precaución al prescribir fenofibrato con colchicina.
Función hepática
El fenofibrato a dosis equivalentes a 107 mg a 160 mg TRICOR por día se ha asociado con aumentos en las transaminasas séricas [AST (SGOT) o ALT (SGPT)]. En un análisis agrupado de 10 ensayos controlados con placebo, se produjo un aumento de> 3 veces el límite superior de la normalidad en el 5,3% de los pacientes que tomaron fenofibrato frente al 1,1% de los pacientes tratados con placebo.
Cuando se siguieron las determinaciones de transaminasas después de la interrupción del tratamiento o durante el tratamiento continuo, generalmente se observó un retorno a los límites normales. La incidencia de aumentos en las transaminasas relacionadas con la terapia con fenofibrato parece estar relacionada con la dosis. En un estudio de 8 semanas para variar la dosis, la incidencia de elevaciones de ALT o AST al menos tres veces el límite superior de la normalidad fue del 13% en pacientes que recibieron dosis equivalentes a 107 mg a 160 mg TRICOR por día y fue del 0% en aquellos que recibieron dosis equivalentes a 54 mg o menos TRICOR por día, o placebo. Se ha informado hepatitis hepatocelular, crónica activa y colestática asociada con la terapia con fenofibrato después de exposiciones de semanas a varios años. En casos extremadamente raros, se ha informado cirrosis en asociación con hepatitis crónica activa.
La monitorización periódica de la línea de base y regular de la función hepática, incluida la ALT sérica (SGPT), debe realizarse durante la terapia con TRICOR, y la terapia debe suspenderse si los niveles de enzimas persisten por encima de tres veces el límite normal.
Creatinina sérica
Se han notificado elevaciones en la creatinina sérica en pacientes con fenofibrato. Estas elevaciones tienden a volver a la línea de base después de la interrupción de fenofibrato. Se desconoce la importancia clínica de estas observaciones. Monitorear la función renal en pacientes con insuficiencia renal que toman TRICOR. La monitorización renal también debe considerarse para pacientes que toman TRICOR en riesgo de insuficiencia renal, como ancianos y pacientes con diabetes.
Colelitiasis
El fenofibrato, como el clofibrato y el gemfibrozilo, puede aumentar la excreción de colesterol en la bilis, lo que lleva a la colelitiasis. Si se sospecha colelitiasis, se indican los estudios de vesícula biliar. La terapia TRICOR debe suspenderse si se encuentran cálculos biliares.
Anticoagulantes de cumarina
Se debe tener precaución cuando los anticoagulantes de cumarina se administran junto con TRICOR debido a la potenciación de los efectos anticoagulantes de tipo cumarina en la prolongación del tiempo de protrombina / relación normalizada internacional (PT / INR). Para prevenir complicaciones hemorrágicas, se recomienda un monitoreo frecuente de PT / INR y un ajuste de dosis del anticoagulante hasta que PT / INR se haya estabilizado.
Pancreatitis
Se ha informado de pancreatitis en pacientes que toman fenofibrato, gemfibrozilo y clofibrato. Esta ocurrencia puede representar un fracaso de eficacia en pacientes con hipertrigliceridemia severa, un efecto farmacológico directo o un fenómeno secundario mediado a través de la formación de piedra del tracto biliar o lodo con obstrucción del conducto biliar común.
Cambios hematológicos
Se han observado disminuciones leves a moderadas de hemoglobina, hematocrito y glóbulos blancos en pacientes después del inicio de la terapia con fenofibrato. Sin embargo, estos niveles se estabilizan durante la administración a largo plazo. Se han notificado trombocitopenia y agranulocitosis en individuos tratados con fenofibrato. Se recomienda la monitorización periódica de los recuentos de glóbulos rojos y blancos durante los primeros 12 meses de administración de TRICOR.
Reacciones de hipersensibilidad
Se han notificado reacciones de hipersensibilidad aguda como el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica que requieren hospitalización del paciente y tratamiento con esteroides en personas tratadas con fenofibratos. Urticaria se vio en 1.1 vs. 0%, y erupción en 1.4 vs. 0.8% de pacientes con fenofibrato y placebo respectivamente en ensayos controlados.
Enfermedad venotromboembólica
En el ensayo FIELD, se observaron embolia pulmonar (EP) y trombosis venosa profunda (TVP) a tasas más altas en el grupo fenofibrato que en el grupo tratado con placebo. De 9.795 pacientes inscritos en FIELD, había 4.900 en el grupo placebo y 4.895 en el grupo fenofibrato. Para DVT, hubo 48 eventos (1%) en el grupo placebo y 67 (1%) en el grupo fenofibrato (p = 0.074); y para PE, hubo 32 (0.7%) eventos en el grupo placebo y 53 (1%) en el grupo fenofibrato (p = 0.022).
En el Proyecto de Medicamentos Coronarios, una mayor proporción del grupo de clofibrato experimentó embolia pulmonar o tromboflebitis mortal o no mortal definida o sospechada que el grupo placebo (5.2% vs. 3.3% a los cinco años; p <0.01).
Disminuciones paradójicas en los niveles de colesterol HDL
Ha habido informes posteriores a la comercialización y ensayos clínicos de disminuciones severas en los niveles de colesterol HDL (tan bajo como 2 mg / dL) en pacientes diabéticos y no diabéticos iniciados en la terapia con fibratos. La disminución en HDL-C se refleja en una disminución en la apolipoproteína A1. Se ha informado que esta disminución ocurre dentro de las 2 semanas a años después del inicio de la terapia con fibrato. Los niveles de HDL-C permanecen deprimidos hasta que se haya retirado la terapia con fibrato; La respuesta a la retirada de la terapia con fibratos es rápida y sostenida. Se desconoce la importancia clínica de esta disminución en HDL-C. Se recomienda verificar los niveles de HDL-C dentro de los primeros meses después del inicio de la terapia con fibrato. Si se detecta un nivel de HDL-C severamente deprimido, se debe retirar la terapia con fibrato y controlar el nivel de HDL-C hasta que haya vuelto al valor inicial, y no se debe reiniciar la terapia con fibrato.
Toxicología no clínica
Carcinogénesis y mutagénesis e deterioro de la fertilidad
Se han realizado dos estudios de carcinogenicidad en la dieta en ratas con fenofibrato. En el primer estudio de 24 meses, las ratas Wistar se dosificaron con fenofibrato a 10, 45 y 200 mg / kg / día, aproximadamente 0.3, 1 y 6 veces la dosis humana máxima recomendada (MRHD), según las comparaciones del área de superficie corporal (mg / metro2). A una dosis de 200 mg / kg / día (a 6 veces el MRHD), la incidencia de carcinomas hepáticos aumentó significativamente en ambos sexos. Se observó un aumento estadísticamente significativo en los carcinomas pancreáticos en los hombres a 1 y 6 veces el MRHD; Se observó un aumento en los adenomas pancreáticos y los tumores de células intersticiales testiculares benignos a 6 veces el MRHD en los hombres. En un segundo estudio de carcinogenicidad en ratas de 24 meses en una cepa diferente de ratas (Sprague-Dawley) dosis de 10 y 60 mg / kg / día (0.3 y 2 veces el MRHD) produjo aumentos significativos en la incidencia de adenomas acinarios pancreáticos en ambos sexos y aumentos en los tumores de células intersticiales testiculares en hombres a 2 veces el MRHD
Se realizó un estudio de carcinogenicidad de 117 semanas en ratas que comparó tres fármacos: fenofibrato 10 y 60 mg / kg / día (0.3 y 2 veces el MRHD), clofibrato (400 mg / kg / día; 2 veces la dosis humana) y gemfibrozilo (250 mg / kg / día;2 superficie). El fenofibrato aumentó los adenomas acinarios pancreáticos en ambos sexos. El clofibrato aumentó el carcinoma hepatocelular y los adenomas acinarios pancreáticos en hombres y los nódulos neoplásicos hepáticos en mujeres. Gemfibrozil aumentó los nódulos neoplásicos hepáticos en hombres y mujeres, mientras que los tres fármacos aumentaron los tumores testiculares de células intersticiales en los hombres.
En un estudio de 21 meses en ratones CF-1, fenofibrato 10, 45 y 200 mg / kg / día (aproximadamente 0.2, 1 y 3 veces el MRHD en base a mg / m2 área de superficie) aumentó significativamente los carcinomas hepáticos en ambos sexos a 3 veces el MRHD. En un segundo estudio de 18 meses a 10, 60 y 200 mg / kg / día, el fenofibrato aumentó significativamente los carcinomas hepáticos en ratones machos y los adenomas hepáticos en ratones hembras a 3 veces el MRHD
Los estudios de microscopía electrónica han demostrado la proliferación peroxisomal después de la administración de fenofibrato a la rata. No se ha realizado un estudio adecuado para evaluar la proliferación de peroxisomas en humanos, pero se han observado cambios en la morfología y los números de peroxisoma en humanos después del tratamiento con otros miembros de la clase de fibrato cuando se compararon las biopsias hepáticas antes y después del tratamiento en el mismo individuo.
Mutagénesis
Se ha demostrado que el fenofibrato carece de potencial mutagénico en las siguientes pruebas: Ames, linfoma de ratón, aberración cromosómica y síntesis de ADN no programada en hepatocitos de rata primaria.
Deterioro de la fertilidad
En los estudios de fertilidad, las ratas recibieron dosis dietéticas orales de fenofibrato, los machos recibieron 61 días antes del apareamiento y las hembras 15 días antes del apareamiento a través del destete, lo que no produjo un efecto adverso sobre la fertilidad a dosis de hasta 300 mg / kg / día (~ 10 veces el MRHD, basado en mg / m2 comparaciones de superficie).
Uso en poblaciones específicas
Embarazo
Embarazo Categoría C
No se ha establecido la seguridad en mujeres embarazadas. No existen estudios adecuados y bien controlados de fenofibrato en mujeres embarazadas. El fenofibrato debe usarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
En ratas hembras que recibieron dosis dietéticas orales de 15, 75 y 300 mg / kg / día de fenofibrato a partir de los 15 días previos al apareamiento a través del destete, se observó toxicidad materna a 0.3 veces el MRHD, según las comparaciones del área de superficie corporal; mg / m2.
En ratas preñadas que recibieron dosis dietéticas orales de 14, 127 y 361 mg / kg / día desde el día de gestación 6-15 durante el período de organogénesis, no se observaron hallazgos adversos del desarrollo a 14 mg / kg / día (menos de 1 veces el MRHD, basado en comparaciones de área de superficie corporal; mg / m2). A múltiplos más altos de dosis humanas se observó evidencia de toxicidad materna.
En conejos preñadas que recibieron dosis de sonda oral de 15, 150 y 300 mg / kg / día desde el día de gestación 618 durante el período de organogénesis y se les permitió administrar, se observaron camadas abortadas a 150 mg / kg / día (10 veces el MRHD, basado en comparaciones de área de superficie corporal: mg / m2). No se observaron hallazgos de desarrollo a 15 mg / kg / día (a menos de 1 veces el MRHD, según las comparaciones del área de superficie corporal; mg / m2).
En ratas preñadas que recibieron dosis dietéticas orales de 15, 75 y 300 mg / kg / día desde el día de gestación 15 hasta el día de lactancia 21 (destete), se observó toxicidad materna a menos de 1 veces la dosis humana máxima recomendada (MRHD), basada en comparaciones de área de superficie corporal; mg / m2.
Madres lactantes
El fenofibrato no debe usarse en madres lactantes. Se debe tomar la decisión de suspender la lactancia o suspender la droga, teniendo en cuenta la importancia de la droga para la madre.
Uso pediátrico
No se ha establecido seguridad ni eficacia en pacientes pediátricos.
Uso geriátrico
Se sabe que el ácido fenofibrico se excreta sustancialmente por el riñón, y el riesgo de reacciones adversas a este medicamento puede ser mayor en pacientes con insuficiencia renal. La exposición al ácido fenofibrico no está influenciada por la edad. Dado que los pacientes de edad avanzada tienen una mayor incidencia de insuficiencia renal, la selección de dosis para los ancianos debe realizarse en función de la función renal. Los pacientes de edad avanzada con función renal normal no deben requerir modificaciones de dosis. Considere controlar la función renal en pacientes de edad avanzada que toman TRICOR
Deterioro renal
Se debe evitar el uso de TRICOR en pacientes con insuficiencia renal grave. Se requiere reducción de la dosis en pacientes con insuficiencia renal leve a moderada. Se recomienda controlar la función renal en pacientes con insuficiencia renal.
Insuficiencia hepática
El uso de TRICOR no se ha evaluado en sujetos con insuficiencia hepática.
EFECTOS ADVERSOS
Experiencia en ensayos clínicos
Debido a que los estudios clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no se pueden comparar directamente con las tasas en los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Los eventos adversos informados por el 2% o más de los pacientes tratados con fenofibrato (y mayor que el placebo) durante los ensayos doble ciego controlados con placebo, independientemente de la causalidad, se enumeran en la Tabla 1 a continuación. Los eventos adversos llevaron a la interrupción del tratamiento en el 5.0% de los pacientes tratados con fenofibrato y en el 3.0% tratados con placebo. Los aumentos en las pruebas de función hepática fueron los eventos más frecuentes, causando la interrupción del tratamiento con fenofibrato en el 1.6% de los pacientes en ensayos doble ciego.
Tabla 1. Reacciones adversas notificadas por el 2% o más de los pacientes tratados con fenofibrato y mayores que el placebo durante los ensayos doble ciego controlados con placebo
| SISTEMA CORPORAL Reacción adversa |
Fenofibrato * | Placebo |
| (N = 439) | (N = 439) | |
| CUERPO COMO TODO | ||
| Dolor abdominal | 4.6% | 4.4% |
| Dolor de espalda | 3.4% | 2.5% |
| Dolor de cabeza | 3.2% | 2.7% |
| DIGESTIVO | ||
| Náuseas | 2.3% | 1.9% |
| Estreñimiento | 2.1% | 1.4% |
| TRASTORNOS METABOLICOS Y NUTRICIONALES | ||
| Pruebas anormales de la función hepática | 7.5% ** | 1.4% |
| Aumento de ALT | 3.0% | 1.6% |
| Aumento de CPK | 3.0% | 1.4% |
| Aumento de AST | 3.4% ** | 0.5% |
| RESPIRACIÓN | ||
| Trastorno respiratorio | 6.2% | 5.5% |
| Rinitis | 2.3% | 1.1% |
| * Dosis equivalente a 160 mg TRICOR ** Significativamente diferente de Placebo. |
||
Experiencia de postmarketing
Se han identificado las siguientes reacciones adversas durante el uso de fenofibrato después de la aprobación: mialgia, rabdomiólisis, pancreatitis, insuficiencia renal aguda, espasmo muscular, hepatitis, cirrosis, anemia, artralgia, disminución de la hemoglobina, disminución del hematocrito, disminución de los glóbulos blancos, astenia y niveles de colesterol HDL severamente deprimidos. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a drogas.
INTERACCIONES DE DROGAS
Anticoagulantes de cumarina
Se ha observado la potenciación de los efectos anticoagulantes de tipo cumarina con la prolongación del PT / INR
Se debe tener precaución cuando se administran anticoagulantes de cumarina junto con TRICOR. La dosis de los anticoagulantes debe reducirse para mantener el PT / INR al nivel deseado para prevenir complicaciones hemorrágicas. Las determinaciones frecuentes de PT / INR son aconsejables hasta que se haya determinado definitivamente que el PT / INR se ha estabilizado.
Inmunosupresores
Los inmunosupesantes como la ciclosporina y el tacrolimus pueden producir nefrotoxicidad con disminuciones en el aclaramiento de creatinina y aumentos en la creatinina sérica, y debido a que la excreción renal es la ruta de eliminación primaria de los fármacos de fibrato, incluido TRICOR, existe el riesgo de que una interacción conduzca al deterioro de la función renal. Los beneficios y riesgos de usar TRICOR (tabletas de fenofibrato) con inmunosupresores y otros agentes potencialmente nefrotóxicos deben considerarse cuidadosamente, y la dosis efectiva más baja empleada y la función renal debe controlarse.
Resinas de unión a ácidos biliares
Dado que las resinas aglutinantes de ácido biliar pueden unirse a otros medicamentos administrados simultáneamente, los pacientes deben tomar TRICOR al menos 1 hora antes o de 4 a 6 horas después de una resina aglutinante de ácido biliar para evitar su absorción.
Colquicina
Se han notificado casos de miopatía, incluida la rabdomiólisis, con fenofibratos administrados conjuntamente con colchicina, y se debe tener precaución al prescribir fenofibrato con colchicina.
Pregnancy Category C
Safety in pregnant women has not been established. There are no adequate and well controlled studies of fenofibrate in pregnant women. Fenofibrate should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In female rats given oral dietary doses of 15, 75, and 300 mg/kg/day of fenofibrate from 15 days prior to mating through weaning, maternal toxicity was observed at 0.3 times the MRHD, based on body surface area comparisons; mg/m2.
In pregnant rats given oral dietary doses of 14, 127, and 361 mg/kg/day from gestation day 6-15 during the period of organogenesis, adverse developmental findings were not observed at 14 mg/kg/day (less than 1 times the MRHD, based on body surface area comparisons; mg/m2). At higher multiples of human doses evidence of maternal toxicity was observed.
In pregnant rabbits given oral gavage doses of 15, 150, and 300 mg/kg/day from gestation day 618 during the period of organogenesis and allowed to deliver, aborted litters were observed at 150 mg/kg/day (10 times the MRHD, based on body surface area comparisons: mg/m2). No developmental findings were observed at 15 mg/kg/day (at less than 1 times the MRHD, based on body surface area comparisons; mg/m2).
In pregnant rats given oral dietary doses of 15, 75, and 300 mg/kg/day from gestation day 15 through lactation day 21 (weaning), maternal toxicity was observed at less than 1 times the maximum recommended human dose (MRHD), based on body surface area comparisons; mg/m2.
Experiencia en ensayos clínicos
Debido a que los estudios clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no se pueden comparar directamente con las tasas en los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
Los eventos adversos informados por el 2% o más de los pacientes tratados con fenofibrato (y mayor que el placebo) durante los ensayos doble ciego controlados con placebo, independientemente de la causalidad, se enumeran en la Tabla 1 a continuación. Los eventos adversos llevaron a la interrupción del tratamiento en el 5.0% de los pacientes tratados con fenofibrato y en el 3.0% tratados con placebo. Los aumentos en las pruebas de función hepática fueron los eventos más frecuentes, causando la interrupción del tratamiento con fenofibrato en el 1.6% de los pacientes en ensayos doble ciego.
Tabla 1. Reacciones adversas notificadas por el 2% o más de los pacientes tratados con fenofibrato y mayores que el placebo durante los ensayos doble ciego controlados con placebo
| SISTEMA CORPORAL Reacción adversa |
Fenofibrato * | Placebo |
| (N = 439) | (N = 439) | |
| CUERPO COMO TODO | ||
| Dolor abdominal | 4.6% | 4.4% |
| Dolor de espalda | 3.4% | 2.5% |
| Dolor de cabeza | 3.2% | 2.7% |
| DIGESTIVO | ||
| Náuseas | 2.3% | 1.9% |
| Estreñimiento | 2.1% | 1.4% |
| TRASTORNOS METABOLICOS Y NUTRICIONALES | ||
| Pruebas anormales de la función hepática | 7.5% ** | 1.4% |
| Aumento de ALT | 3.0% | 1.6% |
| Aumento de CPK | 3.0% | 1.4% |
| Aumento de AST | 3.4% ** | 0.5% |
| RESPIRACIÓN | ||
| Trastorno respiratorio | 6.2% | 5.5% |
| Rinitis | 2.3% | 1.1% |
| * Dosis equivalente a 160 mg TRICOR ** Significativamente diferente de Placebo. |
||
Experiencia de postmarketing
Se han identificado las siguientes reacciones adversas durante el uso de fenofibrato después de la aprobación: mialgia, rabdomiólisis, pancreatitis, insuficiencia renal aguda, espasmo muscular, hepatitis, cirrosis, anemia, artralgia, disminución de la hemoglobina, disminución del hematocrito, disminución de los glóbulos blancos, astenia y niveles de colesterol HDL severamente deprimidos. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a drogas.
There is no specific treatment for overdose with TRICOR. General supportive care of the patient is indicated, including monitoring of vital signs and observation of clinical status, should an overdose occur. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage; usual precautions should be observed to maintain the airway. Because fenofibric acid is highly bound to plasma proteins, hemodialysis should not be considered.
Una variedad de estudios clínicos han demostrado que los niveles elevados de C total, LDL-C y apo B, un complejo de membrana LDL, están asociados con la aterosclerosis humana. Del mismo modo, se asocian niveles disminuidos de HDL-C y su complejo de transporte, la apolipoproteína A (apo AI y apo AII) con el desarrollo de aterosclerosis. Las investigaciones epidemiológicas han establecido que la morbilidad y mortalidad cardiovascular varían directamente con el nivel de C total, LDL-C y TG, e inversamente con el nivel de HDL-C. No se ha determinado el efecto independiente de elevar HDL-C o reducir los triglicéridos (TG) sobre el riesgo de morbilidad y mortalidad cardiovascular.
El ácido fenofibrico, el metabolito activo del fenofibrato, produce reducciones en el colesterol total, el colesterol LDL, la apolipoproteína B, los triglicéridos totales y la lipoproteína rica en triglicéridos (VLDL) en pacientes tratados. Además, el tratamiento con fenofibrato produce aumentos en la lipoproteína de alta densidad (HDL) y las apolipoproteínas apoAI y apoAII
El fenofibrato es un profármaco del ácido fenofibrico de resto químico activo. El fenofibrato se convierte por hidrólisis de éster en el cuerpo en ácido fenofibrico, que es el componente activo medible en la circulación.
Absorción
La biodisponibilidad absoluta de fenofibrato no se puede determinar ya que el compuesto es prácticamente insoluble en medios acuosos adecuados para inyección. Sin embargo, el fenofibrato se absorbe bien del tracto gastrointestinal. Después de la administración oral en voluntarios sanos, aproximadamente el 60% de una dosis única de fenofibrato radiomarcado apareció en la orina, principalmente como ácido fenofibrico y su conjugado de glucuronato, y el 25% se excretó en las heces. Los niveles plasmáticos máximos de ácido fenofibrico ocurren dentro de las 6 a 8 horas posteriores a la administración.
La absorción de fenofibrato aumenta cuando se administra con alimentos. Con las tabletas de fenofibrato, el grado de absorción aumenta aproximadamente un 35% en forma de alimentación en comparación con las condiciones de ayuno.
Distribución
Tras la administración múltiple de fenofibrato, el estado estacionario del ácido fenofíbrico se logra dentro de los 5 días. Las concentraciones plasmáticas de ácido fenofibrico en estado estacionario son aproximadamente el doble de las que siguen a una dosis única. La unión a proteínas séricas fue aproximadamente del 99% en sujetos normales e hiperlipidemicos.
Metabolismo
Después de la administración oral, las esterasas hidrolizan rápidamente fenofibrato al metabolito activo, ácido fenofibrico; no se detecta fenofibrato sin cambios en el plasma.
El ácido fenofibrico se conjuga principalmente con ácido glucurónico y luego se excreta en la orina. Una pequeña cantidad de ácido fenofibrico se reduce en el resto carbonilo a un metabolito benzhidrol que, a su vez, se conjuga con ácido glucurónico y se excreta en la orina.
In vivo los datos del metabolismo indican que ni el fenofibrato ni el ácido fenofibrico sufren un metabolismo oxidativo (p. ej., citocromo P450) en gran medida.
Eliminación
Después de la absorción, el fenofibrato se excreta principalmente en la orina en forma de metabolitos, principalmente ácido fenofibrico y glucurónido de ácido fenofibrico. Después de la administración de fenofibrato radiomarcado, aproximadamente el 60% de la dosis apareció en la orina y el 25% se excretó en las heces.
El ácido fenofibrico se elimina con una vida media de 20 horas, lo que permite una dosificación diaria.
However, we will provide data for each active ingredient





