

Composition:
Application:
Utilisé dans le traitement:
Examiné médicalement par Fedorchenko Olga Valeryevna, Pharmacie Dernière mise à jour le 26.06.2023

Attention! Information sur la page est réservée aux professionnels de la santé! Les informations sont collectées dans des sources ouvertes et peuvent contenir des erreurs significatives! Soyez prudent et revérifiez toutes les informations de cette page!
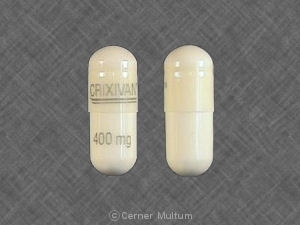
Capsules CRIXIVANES sont fournis comme suit:
No 3756 - 200 mg gélules: blanc semi-translucide. gélules codées «CRIXIVAN ™ 200 mg» en bleu.
Disponible comme:
NDC 0006-0571-43 bouteilles de 360 unités d'utilisation (avec dessiccant).
No 3758 - 400 mg gélules: blanc semi-translucide. gélules codées «CRIXIVAN ™ 400 mg» en vert. Disponible comme:
NDC 0006-0573-62 bouteilles de 180 unités d'utilisation (avec dessiccant)
Stockage
Bouteilles: Conserver dans un récipient hermétiquement fermé à température ambiante, 15-30 ° C (59-86 ° F). Protéger de l'humidité.
Les capsules CRIXIVAN sont sensibles à l'humidité. CRIXIVAN doit être distribué et stocké dans le récipient d'origine. Le dessiccant devrait rester dans la bouteille d'origine.
Dist. par: Merck Sharp & Dohme Corp., une filiale de MERCK & CO., INC., Whitehouse Station, NJ 08889, États-Unis. Révisé le 03/2015
CRIXIVAN en association avec des agents antirétroviraux est indiqué pour le traitement de l'infection par le VIH. Cette indication est basée sur deux essais cliniques d'une durée d'environ 1 an démontrant: 1) une réduction dans le risque de maladies ou de décès définissant le SIDA; 2) une suppression prolongée de ARN VIH .
La posologie recommandée de CRIXIVAN est de 800 mg (généralement deux Capsules de 400 mg) par voie orale toutes les 8 heures.
CRIXIVAN doit être pris à des intervalles de 8 heures. Pour absorption optimale, CRIXIVAN doit être administré sans nourriture mais avec de l'eau 1 heure avant ou 2 heures après un repas. Alternativement, CRIXIVAN peut l'être administré avec d'autres liquides tels que le lait écrémé, le jus, le café ou le thé, ou avec un repas léger, par ex., pain grillé sec avec de la gelée, du jus et du café avec du lait écrémé et sucre; ou des flocons de maïs, du lait écrémé et du sucre. (Voir PHARMACOLOGIE CLINIQUE, Effet des aliments sur l'absorption orale.)
Pour assurer une hydratation adéquate, il est recommandé cela les adultes boivent au moins 1,5 litre (environ 48 onces) de liquides pendant le cours des 24 heures.
Thérapie concomitante (Voir PHARMACOLOGIE CLINIQUE, Interactions médicamenteuses, et / ou PRÉCAUTIONS: INTERACTIONS DE DROGUES.)
Delavirdine
La réduction de la dose de CRIXIVAN à 600 mg toutes les 8 heures devrait être pris en compte lors de l'administration de de la delavirdine 400 mg trois fois par jour.
Didanosine
Si l'indinavir et la didanosine sont administrés en concomitance, ils doivent être administrés à au moins une heure d'intervalle à l'état vide estomac (consulter la circulaire du produit du fabricant pour la didanosine).
Itraconazole
La réduction de la dose de CRIXIVAN à 600 mg toutes les 8 heures est recommandé lors de l'administration simultanée d'itraconazole 200 mg deux fois par jour.
Kétoconazole
La réduction de la dose de CRIXIVAN à 600 mg toutes les 8 heures est recommandé lors de l'administration simultanée de kétoconazole.
Rifabutine
Réduction de la dose de rifabutine à la moitié de la dose standard (consulter la circulaire produit du fabricant pour la rifabutine) et une augmentation de dose de CRIXIVAN à 1000 mg toutes les 8 heures sont recommandés lorsque la rifabutine et CRIXIVAN sont co-administrés.
Insuffisance hépatique
La posologie de CRIXIVAN doit être réduite à 600 mg chacun 8 heures chez les patients présentant une insuffisance hépatique légère à tomodérate due à cirrhose.
Néphrolithiase / Urolithiase
En plus d'une hydratation adéquate, la gestion médicale en les patients qui présentent une néphrolithiase / urolithiase peuvent inclure une temporaire interruption (par ex., 1 à 3 jours) ou arrêt du traitement.
CRIXIVAN est contre-indiqué chez les patients atteints cliniquement hypersensibilité significative à l'un de ses composants.
L'inhibition du CYP3A4 par CRIXIVAN peut entraîner une élévation concentrations plasmatiques des médicaments suivants, pouvant entraîner des effets graves ou réactions mettant la vie en danger:
Tableau 7: Interactions médicamenteuses avec Crixivan:
Médicaments contre-indiqués
| Classe de drogue | Médicaments en classe qui sont contre-indiqués avec CRIXIVAN |
| Antagoniste des récepteurs alpha 1-adrénergiques | alfuzosine |
| Antiarythmiques | amiodarone |
| Dérivés d'ergot | dihydroergotamine, ergonovine, ergotamine, méthylergonovine |
| Agents de motilité GI | cisapride |
| Inhibiteurs de la HMG-CoA Reductase | lovastatine, simvastatine |
| Neuroleptiques | pimozide |
| Inhibiteurs PDE5 | Revatio (sildénafil) [pour le traitement de l'hypertension artérielle pulmonaire] |
| Sédatif / hypnotique | midazolam oral, triazolam, alprazolam |
| * Marque déposée de Pfizer, Inc. |
AVERTISSEMENTS
ALERTE: Découvrez les médicaments qui ne devraient PAS l'être pris avec CRIXIVAN . Cette déclaration est incluse sur la bouteille du produit étiquette.
Néphrolithiase / Urolithiase
Une néphrolithiase / urolithiase s'est produite avec CRIXIVAN thérapie. La fréquence cumulée de la néphrolithiase est sensiblement plus élevée patients pédiatriques (29%) que chez les patients adultes (12,4%; plage à travers l'individu essais: 4,7% à 34,4%). La fréquence cumulée des événements de néphrolithiase augmente avec l'augmentation de l'exposition à CRIXIVAN; cependant, le risque dans le temps reste relativement constant. Dans certains cas, la néphrolithiase / urolithiase l'a fait été associé à une insuffisance rénale ou à une insuffisance rénale aiguë, pyélonéphrite avec ou sans bactérémie. Si signes ou symptômes de une néphrolithiase / urolithiase survient (y compris une douleur au flanc, avec ou sans hématurie ou hématurie microscopique), interruption temporaire (par ex., 1-3 jours) ou l'arrêt du traitement peut être envisagé. Une hydratation adéquate est recommandé chez tous les patients traités par CRIXIVAN. (Voir RÉACTIONS INDÉSIRABLES et DOSAGE ET ADMINISTRATION, Néphrolithiase / Urolithiase.)
Anémie hémolytique
Anémie hémolytique aiguë, y compris les cas entraînant décès, a été rapporté chez des patients traités par CRIXIVAN. Une fois le diagnostic posé des mesures apparentes et appropriées pour le traitement de l'anémie hémolytique devraient être institué, y compris l'arrêt de CRIXIVAN .
Hépatite
Hépatite, y compris les cas entraînant une insuffisance hépatique et le décès a été signalé chez des patients traités par CRIXIVAN. Parce que le la majorité de ces patients avaient des conditions médicales confondantes et / ou recevaient thérapie (s) concomitante (s), une relation causale entre CRIXIVAN et ceux-ci les événements n'ont pas été établis.
Hyperglycémie
Nouveau diabète sucré, exacerbation de la préexistence le diabète sucré et l'hyperglycémie ont été rapportés après la commercialisation surveillance chez les patients infectés par le VIH recevant un traitement par inhibiteur de protéase. Certains patients ont dû initier ou ajuster la dose d'insuline ou par voie orale agents hypoglycémiques pour le traitement de ces événements. Dans certains cas, diabétique une acidocétose s'est produite. Chez les patients qui ont arrêté la protéase thérapie par inhibiteur, l'hyperglycémie a persisté dans certains cas. Parce que ces événements ont été signalés volontairement au cours de la pratique clinique, des estimations de fréquence ne peut pas être fait et une relation causale entre le traitement par inhibiteur de protéase et ces événements n'ont pas été établis.
Risque d'effets indésirables graves dus au médicament Interactions
Initiation de CRIXIVAN, un inhibiteur du CYP3A, chez les patients recevoir des médicaments métabolisés par le CYP3A ou initiation de médicaments métabolisé par le CYP3A chez les patients recevant déjà CRIXIVAN, peut augmenter le plasma concentrations de médicaments métabolisés par le CYP3A. Initiation des médicaments qui inhibent ou induisent le CYP3A peut augmenter ou diminuer les concentrations de CRIXIVAN, respectivement. Ces interactions peuvent conduire à:
- Effets indésirables cliniquement significatifs, potentiellement conduisant à des événements graves, mettant la vie en danger ou mortels dus à une plus grande exposition de médicaments concomitants.
- Effets indésirables cliniquement significatifs d'une augmentation expositions de CRIXIVAN .
- Perte d'effet thérapeutique de CRIXIVAN et possible développement de la résistance.
Voir le tableau 9 pour les étapes permettant de prévenir ou de gérer ces éventuels et les interactions médicamenteuses importantes connues, y compris les recommandations posologiques. Considérez le potentiel d'interactions médicamenteuses avant et pendant CRIXIVAN thérapie; revoir les médicaments concomitants pendant le traitement par CRIXIVAN; et surveiller pour les effets indésirables associés aux médicaments concomitants.
Utilisation concomitante de CRIXIVAN avec la lovastatine ou la simvastatine est contre-indiquée en raison d'un risque accru de myopathie, y compris rhabdomyolyse. Des précautions doivent être prises si CRIXIVAN est utilisé simultanément avec de l'atorvastatine ou de la rosuvastatine. Titrer l'atorvastatine et la rosuvastatine soigneusement et utilisez la dose nécessaire la plus faible avec CRIXIVAN. (Voir PRÉCAUTIONS: INTERACTIONS DE DROGUES.)
Le midazolam est largement métabolisé par le CYP3A4. La co-administration avec CRIXIVAN avec ou sans ritonavir peut provoquer une importante augmentation de la concentration de cette benzodiazépine. Aucune étude d'interaction médicamenteuse a été réalisée pour la co-administration de CRIXIVAN avec des benzodiazépines. Sur la base des données d'autres inhibiteurs du CYP3A4, les concentrations plasmatiques de midazolam devraient être significativement plus élevés lorsque le midazolam est administré par voie orale. CRIXIVAN ne doit donc pas être co-administré avec une administration orale midazolam (voir CONTRAINDICATIONS), alors que la prudence est de mise co-administration de CRIXIVAN et du midazolam parentéral. Données concomitantes l'utilisation du midazolam parentéral avec d'autres inhibiteurs de protéase suggère une possibilité Augmentation de 3 à 4 fois des taux plasmatiques de midazolam. Si CRIXIVAN avec ou sans le ritonavir est co-administré avec le midazolam parentéral, il doit être fait dans un réglage qui assure une surveillance clinique étroite et une médecine appropriée prise en charge en cas de dépression respiratoire et / ou de sédation prolongée. Posologie une réduction du midazolam doit être envisagée, surtout si plus d'un seul la dose de midazolam est administrée.
Une prudence particulière doit être portée lors de la prescription sildénafil, tadalafil ou vardénafil chez les patients recevant de l'indinavir. La co-administration de CRIXIVAN avec ces médicaments devrait se faire augmenter considérablement les concentrations plasmatiques de sildénafil, de tadalafil et vardénafil et peut entraîner une augmentation des événements indésirables, y compris hypotension, changements visuels et priapisme, associés au sildénafil tadalafil et vardénafil (voir CONTRAINDICATIONS et PRÉCAUTIONS: INTERACTIONS DE DROGUES et INFORMATIONS PATIENTES, et le les informations de prescription complètes du fabricant pour le sildénafil, le tadalafil ou vardénafil).
Utilisation concomitante de CRIXIVAN et St. Millepertuis (Hypericum perforatum) ou des produits contenant St. Le millepertuis n'est pas recommandé. Co-administration de CRIXIVAN et St. Il a été démontré que le moût de John diminue considérablement concentrations d'indinavir (voir PHARMACOLOGIE CLINIQUE, MÉDICAMENT INTERACTIONS) et peut entraîner une perte de réponse virologique et possible résistance à CRIXIVAN ou à la classe des inhibiteurs de protéase.
PRÉCAUTIONS
Général
L'hyperbilirubinémie indirecte s'est produite fréquemment pendant le traitement par CRIXIVAN et a rarement été associé à augmentation des transaminases sériques (voir aussi RÉACTIONS INDÉSIRABLES, Clinique Essais et Expérience post-commercialisation). On ne sait pas si CRIXIVAN aggravera l'hyperbilirubinémie physiologique observée chez les nouveau-nés. (Voir Grossesse.)
Néphrite tubulo-interstitielle
Rapports de néphrite tubulo-interstitielle avec médullaire une calcification et une atrophie corticale ont été observées chez les patients atteints leucocyturie sévère asymptomatique (> 100 cellules / champ de puissance élevée). Les patients avec une leucocyturie sévère asymptomatique doit être suivie de près et surveillée fréquemment avec des urinoirs. Une évaluation diagnostique plus approfondie peut être justifiée, et l'arrêt de CRIXIVAN doit être envisagé chez tous les patients présentant une leucocyturie.
Un syndrome de reconstitution immunitaire a été signalé patients traités par un traitement antirétroviral combiné, y compris CRIXIVAN Pendant la phase initiale du traitement antirétroviral combiné, les patients dont les réponses du système immunitaire peuvent développer une réponse inflammatoire à indolent ou infections opportunistes résiduelles (telles que Mycobacterium avium infection, cytomégalovirus, Pneumocystis jirovecii pneumonie [PCP], ou tuberculose), ce qui peut nécessiter une évaluation et un traitement supplémentaires. Auto-immune troubles (tels que la maladie de Graves, la polymyosite et le syndrome de Guillain-Barré) aurait également eu lieu dans le cadre de la reconstitution immunitaire; cependant le délai d'apparition est plus variable et peut se produire plusieurs mois après l'initiation de traitement.
Conditions coexistantes
Patients hémophiles: Il y a eu des rapports de saignement spontané chez les patients atteints d'hémophilie A et B traités avec inhibiteurs de protéase. Chez certains patients, un facteur VIII supplémentaire était nécessaire. Dans de nombreux cas signalés, le traitement par des inhibiteurs de protéase a été poursuivi ou redémarré. Une relation causale entre la thérapie par inhibiteur de protéase et ceux-ci les épisodes n'ont pas été établis. (Voir RÉACTIONS INDÉSIRABLES, Post-marketing Expérience.)
Patients atteints d'insuffisance hépatique due à une cirrhose: Chez ces patients, la posologie de CRIXIVAN doit être abaissée en raison de diminution du métabolisme de CRIXIVAN (voir DOSAGE ET ADMINISTRATION). Les patients avec insuffisance rénale: les patients atteints d'insuffisance rénale ne l'ont pas été étudié.
Redistribution des graisses
Redistribution / accumulation de graisse corporelle, y compris centrale obésité, hypertrophie dorsocervicale des graisses (bosse de buffle), dépérissement périphérique , l'émaciation faciale, l'élargissement mammaire et «l'aspect cushingoïde» l'ont été observé chez les patients recevant un traitement antirétroviral. Le mécanisme et les conséquences à long terme de ces événements sont actuellement inconnues. Un causal aucune relation n'a été établie.
Informations pour les patients
Une déclaration aux patients et aux prestataires de soins de santé est inclus sur l'étiquette de la bouteille du produit. ALERTE: Découvrez les médicaments qui ne doit PAS être pris avec CRIXIVAN . Un insert de paquet patient (PPI) pour CRIXIVAN est disponible pour l'information des patients.
CRIXIVAN n'est pas un remède contre l'infection par le VIH-1 et les patients peut continuer à souffrir de maladies associées à l'infection par le VIH-1, y compris infections opportunistes. Les patients doivent rester sous la garde d'un médecin lors de l'utilisation de CRIXIVAN .
Les patients doivent être avisés d'éviter de faire des choses qui peuvent propager l'infection par le VIH-1 à d'autres.
- Ne partagez pas d'aiguilles ou d'autres équipements d'injection.
- Ne partagez pas d'objets personnels qui peuvent avoir du sang ou les fluides corporels dessus, comme les brosses à dents et les lames de rasoir.
- N'ayez aucune sorte de sexe sans protection. Toujours pratiquer des relations sexuelles sans risque en utilisant un préservatif en latex ou en polyuréthane pour réduire les chances de contact sexuel avec du sperme, des sécrétions vaginales ou du sang.
- N'allaitez pas. Nous ne savons pas si CRIXIVAN le peut être transmis à votre bébé dans votre lait maternel et s'il pourrait nuire à votre bébé. De plus, les mères séropositives ne doivent pas allaiter car le VIH-1 peut être transmis le bébé dans le lait maternel.
Il faut conseiller aux patients de rester sous la garde d'un médecin lors de l'utilisation de CRIXIVAN et ne doit pas modifier ou interrompre le traitement sans consulter d'abord le médecin. Par conséquent, si une dose est oubliée, les patients doit prendre la dose suivante à l'heure prévue et ne doit pas doubler cette dose. La thérapie avec CRIXIVAN doit être initiée et maintenue au dosage recommandé.
CRIXIVAN peut interagir avec certains médicaments; donc, les patients doivent être avisés de signaler à leur médecin l'utilisation de tout autre médicaments sur ordonnance, sans ordonnance ou produits à base de plantes, en particulier St. John's moût.
Pour une absorption optimale, CRIXIVAN doit être administré sans nourriture mais avec de l'eau 1 heure avant ou 2 heures après un repas. Alternativement, CRIXIVAN peut être administré avec d'autres liquides tels que le lait écrémé jus, café ou thé, ou avec un repas léger, par ex., pain grillé sec avec de la gelée, du jus , et café avec lait écrémé et sucre; ou flocons de maïs, lait écrémé et sucre (voir PHARMACOLOGIE CLINIQUE, Effet des aliments sur l'absorption orale et DOSAGE ET ADMINISTRATION). Ingestion de CRIXIVAN avec un repas riche en calories, en matières grasses , et les protéines réduisent l'absorption de l'indinavir.
Patients recevant une phosphodiestérase de type 5 (PDE5) l'inhibiteur (sildénafil, tadalafil ou vardénafil) doit être informé qu'ils peut présenter un risque accru d'événements indésirables associés aux inhibiteurs de la PDE5, y compris hypotension, changements visuels et priapisme, et devrait en signaler rapidement symptômes à leurs médecins (voir CONTRAINDICATIONS et AVERTISSEMENTS, MÉDICAMENT INTERACTIONS).
Les patients doivent être informés de cette redistribution ou une accumulation de graisse corporelle peut survenir chez les patients recevant un traitement antirétroviral et que la cause et les effets à long terme sur la santé de ces conditions ne sont pas connus à ce moment.
Les capsules CRIXIVAN sont sensibles à l'humidité. Les patients doit être informé que CRIXIVAN doit être stocké et utilisé dans l'original le récipient et le dessiccant doivent rester dans la bouteille.
Cancérogenèse, mutagenèse, altération de la fertilité
Des études de cancérogénicité ont été menées chez la souris et le rat. Chez la souris, aucune incidence accrue de type de tumeur n'a été observée. Le plus haut la dose testée chez le rat était de 640 mg / kg / jour; à cette dose, une statistiquement significative une incidence accrue d'adénomes thyroïdiens n'a été observée que chez les rats mâles. À cela dose, l'exposition systémique quotidienne chez le rat était environ 1,3 fois supérieure à exposition systémique quotidienne chez l'homme. Aucun signe de mutagénicité ou de génotoxicité a été observé dans in vitro tests de mutagenèse microbienne (Ames) in vitro alcalin tests d'élution pour la rupture d'ADN , in vitro et in vivo aberration chromosomique études, et in vitro dosages de mutagenèse sur cellules de mammifères. Pas de traitement des effets sur l'accouplement, la fertilité ou la survie de l'embryon ont été observés chez les rats femelles et aucun effet lié au traitement sur les performances de l'accouplement n'a été observé chez les rats mâles à doses fournissant une exposition systémique comparable ou légèrement supérieure à celle-ci avec la dose clinique. De plus, aucun effet lié au traitement n'a été observé dans la fécondité ou la fertilité des femelles non traitées accouplées aux mâles traités.
Grossesse
Catégorie de grossesse C: Toxicité pour le développement des études ont été réalisées chez le lapin (à des doses allant jusqu'à 240 mg / kg / jour), le chien (à doses allant jusqu'à 80 mg / kg / jour) et les rats (à des doses allant jusqu'à 640 mg / kg / jour). Le plus haut les doses de ces études ont produit des expositions systémiques chez ces espèces comparables ou légèrement supérieur à l'exposition humaine. Pas d'extérieur lié au traitement , des changements viscéraux ou squelettiques ont été observés chez le lapin ou le chien. Non des changements externes ou viscéraux liés au traitement ont été observés chez le rat. Augmentation du traitement par rapport aux témoins de l'incidence des côtes surnuméraires (à des expositions égales ou inférieures à celles chez l'homme) et de côtes cervicales (à des expositions comparable ou légèrement supérieur à ceux des humains) ont été observés chez le rat. Dans les trois espèces, aucun effet lié au traitement sur la survie embryonnaire / fœtale ou des poids fœtaux ont été observés.
Chez le lapin, à une dose maternelle de 240 mg / kg / jour, aucun médicament a été détecté dans le plasma fœtal 1 heure après l'administration. Taux plasmatiques fœtaux 2 les heures après l'administration étaient d'environ 3% des taux plasmatiques maternels. Dans les chiens, à une dose maternelle de 80 mg / kg / jour, étaient des taux plasmatiques fœtaux environ 50% des taux plasmatiques maternels 1 et 2 heures après dosage. Chez le rat, à des doses maternelles de 40 et 640 mg / kg / jour, médicament plasma fœtal les taux étaient d'environ 10 à 15% et de 10 à 20% de la drogue plasmatique maternelle niveaux 1 et 2 heures après l'administration, respectivement.
L'indinavir a été administré aux singes rhésus pendant la troisième trimestre de la grossesse (à des doses allant jusqu'à 160 mg / kg deux fois par jour) et à singes rhésus néonatals (à des doses allant jusqu'à 160 mg / kg deux fois par jour). Lorsqu'il est administré aux nouveau-nés, l'indinavir a provoqué une exacerbation de l'hyperbilirubinémie physiologique transitoire vu chez cette espèce après la naissance; les valeurs sériques de bilirubine étaient approximatives quadruple au-dessus des témoins à 160 mg / kg deux fois par jour. Une exacerbation similaire l'a fait ne pas se produire chez les nouveau-nés après une exposition in utero à l'indinavir pendant le troisième trimestre de la grossesse. Chez les singes rhésus, les taux plasmatiques de médicaments fœtaux étaient environ 1 à 2% des taux plasmatiques maternels environ 1 heure après administration maternelle à 40, 80 ou 160 mg / kg deux fois par jour.
Une hyperbilirubinémie s'est produite pendant le traitement par CRIXIVAN (voir PRÉCAUTIONS et RÉACTIONS INDÉSIRABLES). On ne sait pas si CRIXIVAN administré à la mère pendant la période périnatale va exacerber hyperbilirubinémie physiologique chez les nouveau-nés.
Il n'y a pas d'études adéquates et bien contrôlées patientes enceintes. CRIXIVAN ne doit être utilisé pendant la grossesse que si le le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Une dose de CRIXIVAN de 800 mg toutes les 8 heures (avec zidovudine 200 mg toutes les 8 heures et 150 mg de lamivudine deux fois par jour) a été étudié chez 16 personnes infectées par le VIH patientes enceintes à 14 à 28 semaines de gestation lors de l'inscription (étude PACTG 358). Compte tenu des expositions antepartum sensiblement inférieures observées et limitées données dans cette population de patients, l'utilisation de l'indinavir n'est pas recommandée Patientes enceintes infectées par le VIH (voir PHARMACOLOGIE CLINIQUE, Enceinte Les patients).
Registre de grossesse antirétrovirale
Surveiller les résultats maternels et fœtaux des patientes enceintes exposé à CRIXIVAN, un registre de grossesse antirétrovirale a été créé. Les médecins sont encouragés à enregistrer les patients en composant le 1-800-258-4263.
Mères infirmières
Des études sur des rates allaitantes l'ont démontré l'indinavir est excrété dans le lait. Bien que l'on ne sache pas si CRIXIVAN l'est excrété dans le lait maternel, il existe un potentiel d'effets indésirables indinavir chez les nourrissons allaités. Les mères doivent être invitées à arrêter allaiter s'ils reçoivent CRIXIVAN. Ceci est cohérent avec le recommandation des Centers for Disease Control du service de santé publique des États-Unis et prévention que les mères infectées par le VIH n'allaitent pas leurs nourrissons pour éviter risque de transmission postnatale du VIH .
Utilisation pédiatrique
Le schéma posologique optimal pour l'utilisation de l'indinavir les patients pédiatriques n'ont pas été établis. Une dose de 500 mg / m² tous les huit des heures ont été étudiées dans des études non contrôlées de 70 enfants, de 3 à 18 ans âge. Les profils pharmacocinétiques de l'indinavir à cette dose n'étaient pas comparables aux profils précédemment observés chez les adultes recevant la dose recommandée (voir PHARMACOLOGIE CLINIQUE, Pédiatrique). Bien que la suppression virale l'était observé chez certains des 32 enfants qui ont été suivis pendant ce régime 24 semaines, un taux de néphrolithiase sensiblement plus élevé a été signalé lorsque par rapport aux données historiques des adultes (voir AVERTISSEMENTS, Néphrolithiase / Urolithiase). Médecins envisageant d'utiliser l'indinavir chez des patients pédiatriques sans autre les options d'inhibiteur de protéase doivent être conscientes des données limitées disponibles dans cette population et le risque accru de néphrolithiase.
Utilisation gériatrique
Les études cliniques de CRIXIVAN n'ont pas inclus suffisamment nombre de sujets âgés de 65 ans et plus pour déterminer s'ils répondent différemment des sujets plus jeunes. En général, sélection de doses pour une personne âgée le patient doit être prudent, reflétant la fréquence plus élevée de diminution fonction hépatique, rénale ou cardiaque et de maladie concomitante ou autre médicament thérapie.
(voir aussi CONTRAINDICATIONS, AVERTISSEMENTS, PRÉCAUTIONS: INTERACTIONS DE DROGUES) L'indinavir est un inhibiteur de l'isoforme CYP3A4 du cytochrome P450. Co-administration de CRIXIVAN et les médicaments principalement métabolisés par le CYP3A4 peuvent entraîner une augmentation du plasma concentrations de l'autre médicament, qui pourraient augmenter ou prolonger son traitement effets thérapeutiques et indésirables (voir CONTRAINDICATIONS et AVERTISSEMENTS). Basé sur in vitro données dans les microsomes hépatiques humains, l'indinavir n'inhibe pas CYP1A2, CYP2C9, CYP2E1 et CYP2B6. Cependant, l'indinavir peut être un faible inhibiteur du CYP2D6.
L'indinavir est métabolisé par le CYP3A4. Médicaments qui induisent L'activité du CYP3A4 devrait augmenter la clairance de l'indinavir entraînant une baisse des concentrations plasmatiques d'indinavir. Co-administration de CRIXIVAN et d'autres médicaments qui inhibent le CYP3A4 peuvent diminuer la clairance de l'indinavir et peut entraîner une augmentation des concentrations plasmatiques d'indinavir.
Des études d'interaction médicamenteuse ont été réalisées avec CRIXIVAN et d'autres médicaments susceptibles d'être co-administrés et certains médicaments couramment utilisés comme sondes pour les interactions pharmacocinétiques. Les effets de la co-administration de CRIXIVAN sur l'ASC, la Cmax et la Cmin sont résumées dans le tableau 2 (effet d'autres médicaments activés indinavir) et tableau 3 (effet de l'indinavir sur d'autres médicaments). Pour information concernant les recommandations cliniques, voir le tableau 9 in PRÉCAUTIONS.
Tableau 2: Interactions médicamenteuses: paramètres pharmacocinétiques
pour l'indinavir sous la présence du médicament co-administré (voir PRÉCAUTIONS, tableau
9 pour les modifications recommandées dans la dose ou le régime)
| Médicament co-administré | Dose de médicament co-administré (mg) | Dose de CRIXIVAN (mg) | n | Rapport (avec / sans médicament co-administré) des paramètres pharmacocinétiques de l'indinavir (IC à 90%); Aucun effet = 1,00 |
||
| Cmax | AUC | Cmin | ||||
| Cimétidine | 600 deux fois par jour, 6 jours | 400 dose unique | 12 | 1.07 (0,77, 1,49) |
0,98 (0,81, 1,19) |
0,82 (0,69, 0,99) |
| Clarithromycine | 500 q12h, 7 jours | 800 trois fois par jour, 7 jours | 10 | 1.08 (0,85, 1,38) |
1.19 (1,00, 1,42) |
1.57 (1.16, 2.12) |
| Delavirdine | 400 trois fois par jour | 400 trois fois par jour, 7 jours | 28 | 0,64 * (0,48, 0,86) |
Aucun changement significatif * | 2.18 (1.16, 4.12) |
| Delavirdine | 400 trois fois par jour | 600 trois fois par jour, 7 jours | 28 | Aucun changement significatif | 1,53 (1.07, 2.20) |
3,98 * (2.04, 7.78) |
| Efavirenz † | 600 une fois par jour, 10 jours | 1000 trois fois par jour, 10 jours | 20 | |||
| Après la dose du matin | Aucun changement significatif * | 0,67 * (0,61, 0,74) |
0,61 * (0,49, 0,76) |
|||
| Après la dose de l'après-midi | Aucun changement significatif * | 0,63 * (0,54, 0,74) |
0,48 * (0,43, 0,53) |
|||
| Après la dose du soir | 0,71 * (0,57, 0,89) |
0,54 * (0,46, 0,63) |
0,43 * (0,37, 0,50) |
|||
| Fluconazole † | 400 une fois par jour, 8 jours | 1000 trois fois par jour, 7 jours | 11 | 0,87 (0,72, 1,05) |
0,76 (0,59, 0,98) |
0,90 (0,72, 1,12) |
| Jus de pamplemousse | 8 oz. | 400 dose unique | 10 | 0,65 (0,53, 0,79) |
0,73 (0,60, 0,87) |
0,90 (0,71, 1,15) |
| Isoniazid | 300 une fois par jour le matin, 8 jours | 800 trois fois par jour, 7 jours | 11 | 0,95 (0,88, 1,03) |
0,99 (0,87, 1,13) |
0,89 (0,75, 1,06) |
| Itraconazole | 200 deux fois par jour, 7 jours | 600 trois fois par jour, 7 jours | 12 | 0,78 (0,69, 0,88) |
0,99 * (0,91, 1,06) |
1.49 (1,28, 1,74) |
| Kétoconazole | 400 une fois par jour, 7 jours | 600 trois fois par jour, 7 jours | 12 | 0,69 * (0,61, 0,78) |
0,80 * (0,74, 0,87) |
1.29 (1.11, 1.51) |
| 400 une fois par jour, 7 jours | 400 trois fois par jour, 7 jours | 12 | 0,42 * (0,37, 0,47) |
0,44 * (0,41, 0,48) |
0,73 * (0,62, 0,85) |
|
| Méthadone | 20-60 une fois par jour le matin, 8 jours | 800 trois fois par jour, 8 jours | 10 | Voir le texte ci-dessous pour la discussion de l'interaction. | ||
| Quinidine | 200 dose unique | 400 dose unique | 10 | 0,96 (0,79, 1,18) |
1.07 (0,89, 1,28) |
0,93 (0,73, 1,19) |
| Rifabutine | 150 une fois par jour le matin, 10 jours | 800 trois fois par jour, 10 jours | 14 | 0,80 (0,72, 0,89) |
0,68 (0,60, 0,76) |
0,60 (0,51, 0,72) |
| Rifabutine | 300 une fois par jour le matin, 10 jours | 800 trois fois par jour, 10 jours | 10 | 0,75 (0,61, 0,91) |
0,66 (0,56, 0,77) |
0,61 (0,50, 0,75) |
| Rifampin | 600 une fois par jour le matin, 8 jours | 800 trois fois par jour, 7 jours | 12 | 0,13 (0,08, 0,22) |
0,08 (0,06, 0,11) |
Pas fini |
| Ritonavir | 100 fois par jour, 14 jours | 800 deux fois par jour, 14 jours | 10, 16 ‡ | Voir le texte ci-dessous pour la discussion de l'interaction. | ||
| Ritonavir | 200 deux fois par jour, 14 jours | 800 deux fois par jour, 14 jours | 9, 16 ‡ | Voir le texte ci-dessous pour la discussion de l'interaction. | ||
| Sildénafil | 25 dose unique | 800 trois fois par jour | 6 | Voir le texte ci-dessous pour la discussion de l'interaction. | ||
| St. Millepertuis (Hypericum perforatum, standardisé à 0,3% d'hypericine) | 300 trois fois par jour avec les repas, 14 jours | 800 trois fois par jour | 8 | Non disponible | 0,46 (0,34, 0,58) § |
0,19 (0,06, 0,33) § |
| Stavudine (d4T) † |
40 fois par jour, 7 jours | 800 trois fois par jour, 7 jours | 11 | 0,95 (0,80, 1,11) |
0,95 (0,80, 1,12) |
1.13 (0,83, 1,53) |
| Triméthoprime / sulfaméthoxazole | 800 Triméthoprime / 160 Sulfaméthoxazole q12h, 7 jours | 400 quatre fois par jour, 7 jours | 12 | 1.12 (0,87, 1,46) |
0,98 (0,81, 1,18) |
0,83 (0,72, 0,95) |
| Zidovudine † | 200 trois fois par jour, 7 jours | 1000 trois fois par jour, 7 jours | 12 | 1.06 (0,91, 1,25) |
1.05 (0,86, 1,28) |
1.02 (0,77, 1,35) |
| Zidovudine / Lamivudine (3TC) † |
200/150 trois fois par jour, 7 jours | 800 trois fois par jour, 7 jours | 6, 9¶ | 1.05 (0,83, 1,33) |
1.04 (0,67, 1,61) |
0,98 (0,56, 1,73) |
| Toutes les études d'interaction menées en bonne santé ,
Sujets adultes séronégatifs, sauf indication contraire. * Par rapport à l'indinavir 800 mg trois fois par jour seul. † Étude menée sur des sujets séropositifs. ‡ Comparaison avec les données historiques sur 16 sujets recevant de l'indinavir seul. § IC à 95% . ¶ Conception de groupe parallèle; n pour l'indinavir + médicament co-administré, n pour l'indinavir seul. |
Tableau 3: Interactions médicamenteuses: paramètres pharmacocinétiques
pour le médicament co-administré en présence d'indinavir (voir PRÉCAUTIONS, tableau 9 pour
Modifications recommandées en dose ou en régime)
| Médicament co-administré | Dose de médicament co-administré (mg) | Dose de CRIXIVAN (mg) | n | Rapport (avec / sans CRIXIVAN) des paramètres pharmacocinétiques du médicament co-administré (IC à 90%); Aucun effet = 1,00 | ||
| Cmax | AUC | Cmin | ||||
| Clarithromycine | 500 deux fois par jour, 7 jours | 800 trois fois par jour, 7 jours | 12 | 1.19 (1,02, 1,39) |
1.47 (1,30, 1,65) |
1.97 (1,58, 2,46) n = 11 |
| Efavirenz | 200 une fois par jour, 14 jours | 800 trois fois par jour, 14 jours | 20 | Aucun changement significatif | Aucun changement significatif | -- |
| Éthinyle Estradiol (ORTHO-NOVUM 1/35) * | 35 mcg, 8 jours | 800 trois fois par jour, 8 jours | 18 | 1.02 (0,96, 1,09) |
1.22 (1,15, 1,30) |
1.37 (1,24, 1,51) |
| Isoniazid | 300 une fois par jour le matin, 8 jours | 800 trois fois par jour, 8 jours | 11 | 1.34 (1,12, 1,60) |
1.12 (1,03, 1,22) |
1.00 (0,92, 1,08) |
| Méthadone † | 20-60 une fois par jour le matin, 8 jours | 800 trois fois par jour, 8 jours | 12 | 0,93 (0,84, 1,03) |
0,96 (0,86, 1,06) |
1.06 (0,94, 1,19) |
| Noréthindrone (ORTHO-NOVUM 1/35) * |
1 mcg, 8 jours | 800 trois fois par jour, 8 jours | 18 | 1.05 (0,95, 1,16) |
1.26 (1,20, 1,31) |
1.44 (1,32, 1,57) |
| Rifabutine 150 mg une fois par jour le matin, 11 jours + indinavir contre 300 mg une fois par jour le matin, 11 jours seul | 150 une fois par jour le matin, 10 jours | 800 trois fois par jour, 10 jours | 14 | 1.29 (1,05, 1,59) |
1.54 (1,33, 1,79) |
1,99 (1,71, 2,31) n = 13 |
| 300 une fois par jour le matin, 10 jours | 800 trois fois par jour, 10 jours | dix | 2.34 (1,64, 3,35) |
2.73 (1,99, 3,77) |
3.44 (2,65, 4,46) n = 9 |
|
| Ritonavir | 100 fois par jour, 14 jours | 800 deux fois par jour, 14 jours | 10, 4 ‡ | 1.61 (1.13, 2.29) |
1.72 (1,20, 2,48) |
1.62 (0,93, 2,85) |
| 200 deux fois par jour, 14 jours | 800 deux fois par jour, 14 jours | 9, 5 ‡ | 1.19 (0,85, 1,66) |
1.96 (1,39, 2,76) |
4.71 (2,66, 8,33) n = 9, 4 |
|
| Saquinavir | ||||||
| Formulation de gel dur | 600 doses uniques | 800 trois fois par jour, 2 jours | 6 | 4.7 (2.7, 8.1) |
6.0 (4.0, 9.1) |
2.9 § (1,7, 4,7) |
| Formulation de gel doux Formulation de gel doux | 800 dose unique 1200 dose unique | 800 trois fois par jour, 2 jours 800 trois fois par jour, 2 jours | 6 | 6.5 (4.7, 9.1) |
7.2 (4.3, 11.9) |
5.5 § (2.2, 14.1) |
| 6 | 4.0 (2,7, 5,9) |
4.6 (3.2, 6.7) |
5.5 § (3.7, 8.3) |
|||
| Sildénafil | 25 dose unique | 800 trois fois par jour | 6 | Voir le texte ci-dessous pour la discussion de l'interaction. | ||
| Stavudine¶ | 40 fois par jour, 7 jours | 800 trois fois par jour, 7 jours | 13 | 0,86 (0,73, 1,03) |
1.21 (1,09, 1,33) |
Pas fini |
| Théophylline | 250 doses uniques (aux jours 1 et 7) | 800 trois fois par jour, 6 jours (Jours 2 à 7) |
12, 4 ‡ | 0,88 (0,76, 1,03) |
1.14 (1,04, 1,24) |
1.13 (0,86, 1,49) n = 7, 3 |
| Triméthoprime / sulfaméthoxazole | ||||||
| Triméthoprime | 800 Triméthoprime / 160 Sulfaméthoxazole q12h, 7 jours | 400 q6h, 7 jours | 12 | 1.18 (1,05, 1,32) |
1.18 (1,05, 1,33) |
1.18 (1,00, 1,39) |
| Triméthoprime / sulfaméthoxazole | ||||||
| Sulfaméthoxazole | 800 Triméthoprime / 160 Sulfaméthoxazole q12h, 7 jours | 400 q6h, 7 jours | 12 | 1.01 (0,95, 1,08) |
1.05 (1.01, 1.09) |
1.05 (0,97, 1,14) |
| Vardénafil | 10 dose unique | 800 trois fois par jour | 18 | Voir le texte ci-dessous pour la discussion de l'interaction. | ||
| Zidovudine¶ | 200 trois fois par jour, 7 jours | 1000 trois fois par jour, 7 jours | 12 | 0,89 (0,73, 1,09) |
1.17 (1,07, 1,29) |
1.51 (0,71, 3,20) n = 4 |
| Zidovudine / Lamivudine¶ | ||||||
| Zidovudine | 200/150 trois fois par jour, 7 jours | 800 trois fois par jour, 7 jours | 6, 7 ‡ | 1.23 (0,74, 2,03) |
1.39 (1,02, 1,89) |
1.08 (0,77, 1,50) n = 5, 5 |
| Zidovudine / Lamivudine¶ | ||||||
| Lamivudine | 200/150 trois fois par jour, 7 jours | 800 trois fois par jour, 7 jours | 6, 7 ‡ | 0,73 (0,52, 1,02) |
0,91 (0,66, 1,26) |
0,88 (0,59, 1,33) |
| Toutes les études d'interaction menées en bonne santé ,
Sujets adultes séronégatifs, sauf indication contraire. * Marque déposée d'Ortho Pharmaceutical Corporation. † Étude menée chez des sujets sur l'entretien de la méthadone. ‡ Conception de groupe parallèle; n pour le médicament co-administré + indinavir, n pour médicament co-administré seul. § C6hr ¶ Étude menée sur des sujets séropositifs. |
Delavirdine
La delavirdine inhibe le métabolisme de l'indinavir tel cette co-administration de 400 mg ou 600 mg d'indinavir trois fois par jour avec 400 mg de de delavirdine trois fois par jour modifie l'ASC, la Cmax et la Cmin de l'indinavir (voir tableau 2). L'indinavir n'a eu aucun effet sur la pharmacocinétique de la delavirdine (voir DOSAGE ET ADMINISTRATION, Thérapie concomitante, Delavirdine), basé sur une comparaison avec données pharmacocinétiques historiques de la delavirdine.
Méthadone
Administration d'indinavir (800 mg toutes les 8 heures) avec méthadone (20 mg à 60 mg par jour) pendant une semaine chez les sujets sous méthadone l'entretien n'a entraîné aucun changement dans l'ASC de la méthadone. Basé sur une comparaison avec données historiques, il y a eu peu ou pas de changement dans l'ASC de l'indinavir
Ritonavir
Comparé aux données historiques des patients qui ont reçu indinavir 800 mg toutes les 8 heures seul, co-administration biquotidienne à des volontaires d'indinavir 800 mg et de ritonavir avec de la nourriture pendant deux semaines ont résulté dans une augmentation de 2,7 fois de l'ASC24h de l'indinavir, une augmentation de 1,6 fois de la Cmax de l'indinavir et une augmentation de 11 fois de l'indinavir Cmin pour une dose de 100 mg de ritonavir et a Augmentation de 3,6 fois de l'ASC24h de l'indinavir, une augmentation de 1,8 fois de la Cmax de l'indinavir et une augmentation de 24 fois de l'indinavir Cmin pour une dose de ritonavir de 200 mg. Dans le même étude, co-administration biquotidienne de l'indinavir (800 mg) et du ritonavir (100 ou 200 mg) a entraîné une augmentation de l'ASC24h du ritonavir par rapport aux mêmes doses de ritonavir seul (voir tableau 3).
Sildénafil
Les résultats d'une étude publiée chez des hommes infectés par le VIH (n = 6) ont indiqué que la co-administration de l'indinavir (800 mg toutes les 8 heures chroniquement) avec une dose unique de 25 mg de sildénafil a entraîné une augmentation de 11% en moyenne 0 à 8 heures d'indinavir et une augmentation de 48% du pic moyen d'indinavir concentration (Cmax) comparée à 800 mg toutes les 8 heures seulement. Sildénafil moyen L'ASC a augmenté de 340% après la co-administration de sildénafil et indinavir par rapport aux données historiques après l'administration de sildénafil seul (voir CONTRAINDICATIONS, AVERTISSEMENTS, INTERACTIONS DE DROGUES et PRÉCAUTIONS: INTERACTIONS DE DROGUES).
Vardénafil
Indinavir (800 mg toutes les 8 heures) co-administré avec a une dose unique de 10 mg de vardénafil a entraîné une augmentation de 16 fois du vardénafil AUC, une augmentation de 7 fois de la Cmax du vardénafil et une augmentation de 2 fois du vardénafil demi-vie (voir AVERTISSEMENTS, INTERACTIONS DE DROGUES et PRÉCAUTIONS: INTERACTIONS DE DROGUES).
Catégorie de grossesse C: Toxicité pour le développement des études ont été réalisées chez le lapin (à des doses allant jusqu'à 240 mg / kg / jour), le chien (à doses allant jusqu'à 80 mg / kg / jour) et les rats (à des doses allant jusqu'à 640 mg / kg / jour). Le plus haut les doses de ces études ont produit des expositions systémiques chez ces espèces comparables ou légèrement supérieur à l'exposition humaine. Pas d'extérieur lié au traitement , des changements viscéraux ou squelettiques ont été observés chez le lapin ou le chien. Non des changements externes ou viscéraux liés au traitement ont été observés chez le rat. Augmentation du traitement par rapport aux témoins de l'incidence des côtes surnuméraires (à des expositions égales ou inférieures à celles chez l'homme) et de côtes cervicales (à des expositions comparable ou légèrement supérieur à ceux des humains) ont été observés chez le rat. Dans les trois espèces, aucun effet lié au traitement sur la survie embryonnaire / fœtale ou des poids fœtaux ont été observés.
Chez le lapin, à une dose maternelle de 240 mg / kg / jour, aucun médicament a été détecté dans le plasma fœtal 1 heure après l'administration. Taux plasmatiques fœtaux 2 les heures après l'administration étaient d'environ 3% des taux plasmatiques maternels. Dans les chiens, à une dose maternelle de 80 mg / kg / jour, étaient des taux plasmatiques fœtaux environ 50% des taux plasmatiques maternels 1 et 2 heures après dosage. Chez le rat, à des doses maternelles de 40 et 640 mg / kg / jour, médicament plasma fœtal les taux étaient d'environ 10 à 15% et de 10 à 20% de la drogue plasmatique maternelle niveaux 1 et 2 heures après l'administration, respectivement.
L'indinavir a été administré aux singes rhésus pendant la troisième trimestre de la grossesse (à des doses allant jusqu'à 160 mg / kg deux fois par jour) et à singes rhésus néonatals (à des doses allant jusqu'à 160 mg / kg deux fois par jour). Lorsqu'il est administré aux nouveau-nés, l'indinavir a provoqué une exacerbation de l'hyperbilirubinémie physiologique transitoire vu chez cette espèce après la naissance; les valeurs sériques de bilirubine étaient approximatives quadruple au-dessus des témoins à 160 mg / kg deux fois par jour. Une exacerbation similaire l'a fait ne pas se produire chez les nouveau-nés après une exposition in utero à l'indinavir pendant le troisième trimestre de la grossesse. Chez les singes rhésus, les taux plasmatiques de médicaments fœtaux étaient environ 1 à 2% des taux plasmatiques maternels environ 1 heure après administration maternelle à 40, 80 ou 160 mg / kg deux fois par jour.
Une hyperbilirubinémie s'est produite pendant le traitement par CRIXIVAN (voir PRÉCAUTIONS et RÉACTIONS INDÉSIRABLES). On ne sait pas si CRIXIVAN administré à la mère pendant la période périnatale va exacerber hyperbilirubinémie physiologique chez les nouveau-nés.
Il n'y a pas d'études adéquates et bien contrôlées patientes enceintes. CRIXIVAN ne doit être utilisé pendant la grossesse que si le le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Une dose de CRIXIVAN de 800 mg toutes les 8 heures (avec zidovudine 200 mg toutes les 8 heures et 150 mg de lamivudine deux fois par jour) a été étudié chez 16 personnes infectées par le VIH patientes enceintes à 14 à 28 semaines de gestation lors de l'inscription (étude PACTG 358). Compte tenu des expositions antepartum sensiblement inférieures observées et limitées données dans cette population de patients, l'utilisation de l'indinavir n'est pas recommandée Patientes enceintes infectées par le VIH (voir PHARMACOLOGIE CLINIQUE, Enceinte Les patients).
Registre de grossesse antirétrovirale
Surveiller les résultats maternels et fœtaux des patientes enceintes exposé à CRIXIVAN, un registre de grossesse antirétrovirale a été créé. Les médecins sont encouragés à enregistrer les patients en composant le 1-800-258-4263.
Essais cliniques chez les adultes
Néphrolithiase / urolithiase, y compris douleur au flanc avec ou sans hématurie (y compris hématurie microscopique), a été signalé dans environ 12,4% (301/2429; plage dans les essais individuels: 4,7% à 34,4%) des patients recevant CRIXIVAN à la dose recommandée dans les essais cliniques avec un suivi médian de 47 semaines (intervalle: 1 jour à 242 semaines; 2238 patients-années suivi). La fréquence cumulée des événements de néphrolithiase augmente avec durée d'exposition à CRIXIVAN; cependant, le risque dans le temps demeure relativement constant. Parmi les patients traités par CRIXIVAN qui se sont développés néphrolithiase / urolithiase dans les essais cliniques pendant la phase en double aveugle, 2,8% (7/246) auraient développé une hydronéphrose et 4,5% (11/246) a subi un placement de stent. Après l'épisode aigu, 4,9% (12/246) de les patients ont arrêté le traitement. (Voir AVERTISSEMENTS et DOSAGE ET ADMINISTRATION, Néphrolithiase / Urolithiase.)
Hyperbilirubinémie asymptomatique (bilirubine totale ≥ 2,5 mg / dL), rapportée principalement sous forme de bilirubine indirecte élevée, a survenu chez environ 14% des patients traités par CRIXIVAN. Dans <1% de cela était associé à des élévations de l'ALT ou de l'AST
Hyperbilirubinémie et néphrolithiase / urolithiasie est survenu plus fréquemment à des doses supérieures à 2,4 g / jour par rapport aux doses ≤ 2,4 g / jour.
Expériences indésirables cliniques rapportées dans ≥ 2% des cas patients traités par CRIXIVAN seul, CRIXIVAN en association avec la zidovudine ou zidovudine plus lamivudine, zidovudine seule ou zidovudine plus lamivudine sont présentés dans le tableau 10.
Tableau 10: Expériences cliniques indésirables rapportées dans ≥ 2%
des patients
| Expérience indésirable | Étude 028 Considéré comme lié aux drogues et d'intensité modérée ou sévère | Étude ACTG 320 de la relation médicamenteuse inconnue et de l'intensité sévère ou mortelle | |||
| Pourcentage CRIXIVAN (n = 332) |
CRIXIVAN plus Zidovudine Pourcentage (n = 332) |
Pourcentage de zidovudine (n = 332) |
CRIXIVAN plus Zidovudine plus Lamivudine Pourcentage (n = 571) |
Pourcentage de zidovudine plus lamivudine (n = 575) |
|
| Corps dans son ensemble | |||||
| Douleur abdominale | 16.6 | 16.0 | 12.0 | 1.9 | 0,7 |
| Asthénie / fatigue | 2.1 | 4.2 | 3.6 | 2.4 | 4.5 |
| Fièvre | 1.5 | 1.5 | 2.1 | 3.8 | 3.0 |
| Malaise | 2.1 | 2.7 | 1.8 | 0 | 0 |
| Système digestif | |||||
| Nausées | 11.7 | 31.9 | 19.6 | 2.8 | 1.4 |
| Diarrhée | 3.3 | 3.0 | 2.4 | 0,9 | 1.2 |
| Vomissements | 8.4 | 17.8 | 9.0 | 1.4 | 1.4 |
| Régurgitation acide | 2.7 | 5.4 | 1.8 | 0,4 | 0 |
| Anorexie | 2.7 | 5.4 | 3.0 | 0,5 | 0,2 |
| Augmentation de l'appétit | 2.1 | 1.5 | 1.2 | 0 | 0 |
| Dyspepsie | 1.5 | 2.7 | 0,9 | 0 | 0 |
| Jaunisse | 1.5 | 2.1 | 0,3 | 0 | 0 |
| Système hémicolique et lymphatique | |||||
| Anémie | 0,6 | 1.2 | 2.1 | 2.4 | 3.5 |
| Système musculo-squelettique | |||||
| Douleurs au dos | 8.4 | 4.5 | 1.5 | 0,9 | 0,7 |
| Système nerveux / psychiatrique | |||||
| Maux de tête | 5.4 | 9.6 | 6.0 | 2.4 | 2.8 |
| Vertiges | 3.0 | 3.9 | 0,9 | 0,5 | 0,7 |
| Somnolence | 2.4 | 3.3 | 3.3 | 0 | 0 |
| Peau et peau | |||||
| Prurit | 4.2 | 2.4 | 1.8 | 0,5 | 0 |
| Éruption cutanée | 1.2 | 0,6 | 2.4 | 1.1 | 0,5 |
| Système respiratoire | |||||
| Toux | 1.5 | 0,3 | 0,6 | 1.6 | 1.0 |
| Difficulté à respirer / dyspnée / essoufflement | 0 | 0,6 | 0,3 | 1.8 | 1.0 |
| Système urogénital | |||||
| Néphrolithiase / urolithiase | 8.7 | 7.8 | 2.1 | 2.6 | 0,3 |
| Dysurie | 1.5 | 2.4 | 0,3 | 0,4 | 0,2 |
| Sens spéciaux | |||||
| Perversion gustative | 2.7 | 8.4 | 1.2 | 0,2 | 0 |
| * Y compris les coliques rénales, et douleur au flanc avec et sans hématurie | |||||
En phase I et II contrôlés les essais, les événements indésirables suivants ont été rapportés beaucoup plus fréquemment par ceux randomisés dans les bras contenant CRIXIVAN que par ceux-ci randomisé en analogues nucléosidiques: éruption cutanée, infection des voies respiratoires supérieures, sèche peau, pharyngite, perversion gustative.
Laboratoire sélectionné anomalies de l'intensité sévère ou mortelle rapportées chez les patients traité avec CRIXIVAN seul, CRIXIVAN en association avec la zidovudine ou la zidovudine plus lamivudine, la zidovudine seule ou la zidovudine plus lamivudine le sont présenté dans le tableau 11.
Tableau 11: Anomalies de laboratoire sélectionnées de gravité
ou Intensité potentiellement mortelle rapportée dans les études 028 et ACTG 320
| Étude 028 Considéré comme lié aux drogues et d'intensité modérée ou sévère | Étude ACTG 320 de la relation médicamenteuse inconnue et de l'intensité sévère ou mortelle | ||||
| Pourcentage CRIXIVAN (n = 329) |
CRIXIVAN plus Zidovudine Pourcentage (n = 320) |
Pourcentage de zidovudine (n = 330) |
CRIXIVAN plus Zidovudine plus Lamivudine Pourcentage (n = 571) |
Pourcentage de zidovudine plus lamivudine (n = 575) |
|
| Hématologie | |||||
| Hémoglobine diminuée <7,0 g / dL | 0,6 | 0,9 | 3.3 | 2.4 | 3.5 |
| Diminution du nombre de plaquettes <50 THS / mm³ | 0,9 | 0,9 | 1.8 | 0,2 | 0,9 |
| Baisse des neutrophiles <0,75 THS / mm³ | 2.4 | 2.2 | 6.7 | 5.1 | 14.6 |
| Chimie du sang | |||||
| ALT accrue> 500% ULN * | 4.9 | 4.1 | 3.0 | 2.6 | 2.6 |
| AST augmenté> 500% ULN | 3.7 | 2.8 | 2.7 | 3.3 | 2.8 |
| Bilirubine sérique totale> 250% LSN | 11.9 | 9.7 | 0,6 | 6.1 | 1.4 |
| Augmentation de l'amylase sérique> 200% LSN | 2.1 | 1.9 | 1.8 | 0,9 | 0,3 |
| Augmentation du glucose> 250 mg / dL | 0,9 | 0,9 | 0,6 | 1.6 | 1.9 |
| Créatinine accrue> 300% LSN | 0 | 0 | 0,6 | 0,2 | 0 |
| * Limite supérieure de la normale gamme. | |||||
Expérience post-commercialisation
Corps dans son ensemble: redistribution / accumulation de graisse corporelle (voir PRÉCAUTIONS, Redistribution des graisses).
Système cardiovasculaire: troubles cardiovasculaires y compris l'infarctus du myocarde et l'angine de poitrine; trouble cérébrovasculaire.
Système digestif: fonction hépatique anomalies; hépatite, y compris les rapports d'insuffisance hépatique (voir AVERTISSEMENTS); pancréatite; jaunisse; distension abdominale; dyspepsie.
Hématologique: augmenté spontané saignement chez les patients hémophiles (voir PRÉCAUTIONS); hémolytique aigu anémie (voir AVERTISSEMENTS).
Endocrinien / Métabolique: nouveau diabète d'apparition sucré, exacerbation du diabète sucré préexistant, hyperglycémie (voir AVERTISSEMENTS).
Hypersensibilité: réactions anaphylactoïdes; urticaire; vascularite.
Système musculo-squelettique: arthralgie, périarthrite.
Système nerveux / psychiatrique: paresthésie orale; dépression.
Peau et peau Appendice: éruption cutanée, y compris érythème syndrome de Stevens-Johnson; hyperpigmentation; alopécie; incarné ongles et / ou paronychie; prurit.
Système urogénital: néphrolithiase / urolithiase, dans certains cas, entraînant une insuffisance rénale ou une insuffisance rénale aiguë pyélonéphrite avec ou sans bactérémie (voir AVERTISSEMENTS); interstitiel néphrite parfois avec des dépôts cristallins d'indinavir; chez certains patients, le la néphrite interstitielle n'a pas disparu après l'arrêt de CRIXIVAN ; insuffisance rénale; insuffisance rénale; leucocyturie (voir PRÉCAUTIONS), cristallurie; dysurie.
Anomalies de laboratoire
Augmentation des triglycérides sériques; augmentation du cholestérol sérique.
Il y a eu plus de 60 rapports d'aiguilles ou de chroniques surdosage humain (jusqu'à 23 fois la dose quotidienne totale recommandée de 2400 mg) avec CRIXIVAN. Les symptômes les plus fréquemment rapportés étaient rénaux (par ex., néphrolithiase / urolithiasie, douleur au flanc, hématurie) et gastro-intestinale (par ex., nausées, vomissements, diarrhée).
On ne sait pas si CRIXIVAN est dialyzable par péritonéale ou hémodialyse.
Absorption
L'indinavir a été rapidement absorbé à jeun avec un temps jusqu'à la concentration plasmatique maximale (Tmax) de 0,8 ± 0,3 heure (moyenne ± S.D.) (n = 11). Une augmentation supérieure à la dose proportionnelle du plasma indinavir des concentrations ont été observées sur la plage de doses de 200 à 1 000 mg. À un dosage régime de 800 mg toutes les 8 heures, zone à l'état d'équilibre sous le plasma la courbe de concentration dans le temps (ASC) était de 30 691 ± 11 407 nMà ¢ ¬Â ¢ heure (n = 16), plasma de crête la concentration (Cmax) était de 12 617 ± 4037 nM (n = 16) et la concentration plasmatique huit heures après la dose (auge) était de 251 ± 178 nM (n = 16).
Effet des aliments sur l'absorption orale
Administration de l'indinavir avec un repas riche en calories , les graisses et les protéines (784 kcal, 48,6 g de graisses, 31,3 g de protéines) ont entraîné une augmentation de 77% ± 8% réduction de l'ASC et réduction de 84% ± 7% de la Cmax (n = 10). Administration avec des repas plus légers (par ex., un repas de pain grillé sec avec de la gelée, du jus de pomme et du café avec du lait écrémé et du sucre ou un repas de flocons de maïs, de lait écrémé et de sucre) a entraîné peu ou pas de changement dans l'ASC, la Cmax ou la concentration minimale.
Distribution
L'indinavir était lié à environ 60% au plasma humain protéines sur une plage de concentration de 81 nM à 16 300 nM
Métabolisme
Après une dose de 400 mg de 14C-indinavir, 83 ± 1% (n = 4) et 19 ± 3% (n = 6) de la radioactivité totale ont été récupérés dans les excréments et l'urine respectivement; la radioactivité due au médicament parent dans les excréments et l'urine était de 19,1% et 9,4%, respectivement. Sept métabolites ont été identifiés, un conjugué glucuronide et six métabolites oxydants. In vitro des études indiquent que le cytochrome P-450 3A4 (CYP3A4) est la principale enzyme responsable de la formation de l'oxydatif métabolites.
Élimination
Moins de 20% de l'indinavir est excrété sous forme inchangée dans le urine. L'excrétion urinaire moyenne du médicament inchangé était de 10,4 ± 4,9% (n = 10) et de 12,0 ± 4,9% (n = 10) après une dose unique de 700 mg et 1000 mg, respectivement. L'indinavir a été rapidement éliminé avec une demi-vie de 1,8 ± 0,4 heure (n = 10). Aucune accumulation significative n'a été observée après plusieurs doses à 800 mg chacune 8 heures.
Disponible dans les pays
 Australia
Australia Austria
Austria Belgium
Belgium Brazil
Brazil Bulgaria
Bulgaria Canada
Canada China
China Cyprus
Cyprus Czech Republic
Czech Republic Denmark
Denmark Estonia
Estonia Finland
Finland France
France Germany
Germany Greece
Greece Hong Kong
Hong Kong Hungary
Hungary Iceland
Iceland Ireland
Ireland Italy
Italy Japan
Japan Latvia
Latvia Lebanon
Lebanon Liechtenstein
Liechtenstein Lithuania
Lithuania Luxembourg
Luxembourg Malta
Malta Netherlands
Netherlands New Zealand
New Zealand Norway
Norway Philippines
Philippines Poland
Poland Portugal
Portugal Romania
Romania Russia
Russia Serbia
Serbia Slovakia
Slovakia Slovenia
Slovenia South Africa
South Africa Spain
Spain Sweden
Sweden Switzerland
Switzerland Taiwan
Taiwan Turkey
Turkey United Kingdom
United Kingdom USA
USA Venezuela
Venezuela