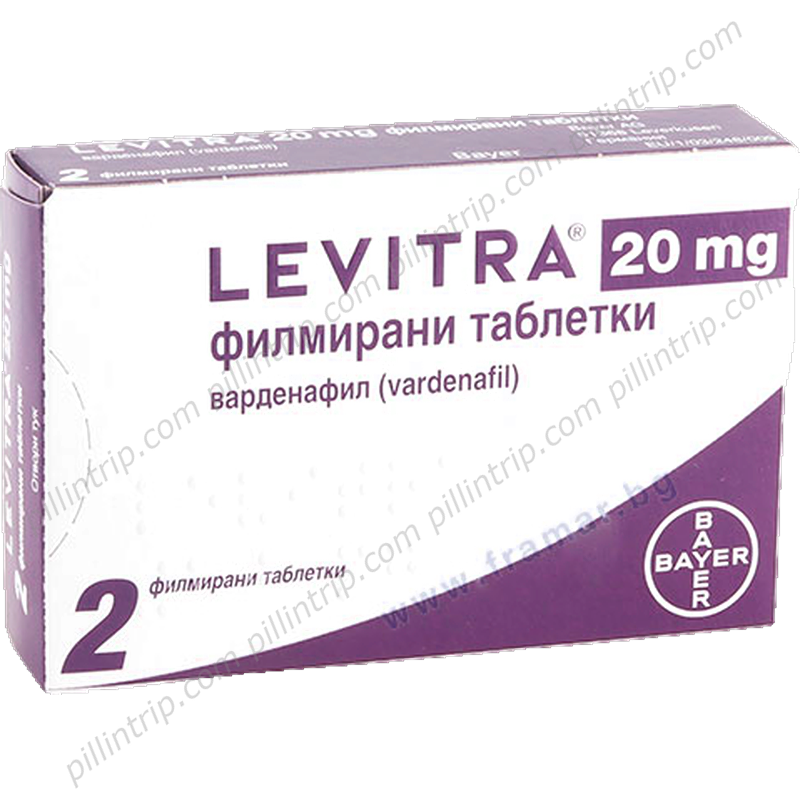
Levitra
Links rápidos para seções importantes
Medically reviewed
Last updated on 12/22/2025
Esta página fornece informações gerais e de referência compiladas a partir de fontes médicas oficiais. Não substitui aconselhamento médico profissional, diagnóstico ou tratamento. Para decisões sobre a sua saúde, consulte um profissional de saúde qualificado.
Visão geral de Levitra
Quais efeitos secundários são possíveis com Levitra?
Sobredosagem e resposta em caso de emergência
Usos terapêuticos de Levitra
Critérios de utilização e restrições
O que devo saber sobre interações com outros medicamentos?
Mecanismo de ação
Informações sobre dosagem e administração
Evidência clínica recente
Perguntas frequentes (FAQ)
Como Levitra deve ser armazenado e descartado?
Atenção! Sempre consulte um médico ou farmacêutico antes de usar pílulas ou medicamentos.
Disponível em países:
![]() Australia
Australia
![]() Austria
Austria
![]() Bahrain
Bahrain
![]() Belgium
Belgium
![]() Bosnia & Herzegowina
Bosnia & Herzegowina
![]() Brasil
Brasil
![]() Bulgaria
Bulgaria
![]() Canada
Canada
![]() Chile
Chile
![]() China
China
![]() Colombia
Colombia
![]() Costa Rica
Costa Rica
![]() Croatia (Hrvatska)
Croatia (Hrvatska)
![]() Cyprus
Cyprus
![]() Czech Republic
Czech Republic
![]() Denmark
Denmark
![]() Ecuador
Ecuador
![]() Egypt
Egypt
![]() Estonia
Estonia
![]() Finland
Finland
![]() France
France
![]() Georgia
Georgia
![]() Germany
Germany
![]() Greece
Greece
![]() Hong Kong
Hong Kong
![]() Hungary
Hungary
![]() Indonesia
Indonesia
![]() Israel
Israel
![]() Italy
Italy
![]() Latvia
Latvia
![]() Lebanon
Lebanon
![]() Lithuania
Lithuania
![]() Malasia
Malasia
![]() Malta
Malta
![]() Mexico
Mexico
![]() Netherlands
Netherlands
![]() New Zealand
New Zealand
![]() Norway
Norway
![]() Oman
Oman
![]() Peru
Peru
![]() Phillipines
Phillipines
![]() Poland
Poland
![]() Portugal
Portugal
![]() Romania
Romania
![]() Russia
Russia
![]() Serbia
Serbia
![]() Slovakia
Slovakia
![]() South Africa
South Africa
![]() Spain
Spain
![]() Sweden
Sweden
![]() Taiwan
Taiwan
![]() Thailand
Thailand
![]() Turkey
Turkey
![]() United Kingdom
United Kingdom
![]() USA
USA
![]() Vietnam
Vietnam
Equivalente a Levitra encontrado em: